

AQP4 Aggravates Cognitive Impairment in Sepsis-Associated Encephalopathy through Inhibiting Nav 1.6-Mediated Astrocyte Autophagy
- PMID: 36922751
- PMCID: PMC10190498
- DOI: 10.1002/advs.202205862
AQP4 Aggravates Cognitive Impairment in Sepsis-Associated Encephalopathy through Inhibiting Nav 1.6-Mediated Astrocyte Autophagy
Erratum in
-
AQP4 Aggravates Cognitive Impairment in Sepsis-Associated Encephalopathy through Inhibiting Nav1.6-Mediated Astrocyte Autophagy.Adv Sci (Weinh). 2023 Oct;10(30):e2306241. doi: 10.1002/advs.202306241. Adv Sci (Weinh). 2023. PMID: 37883742 Free PMC article. No abstract available.
Abstract
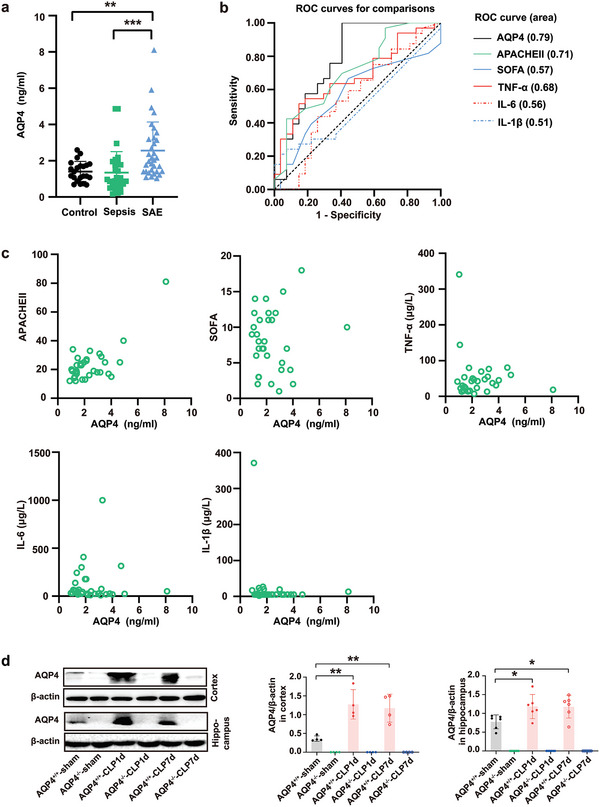
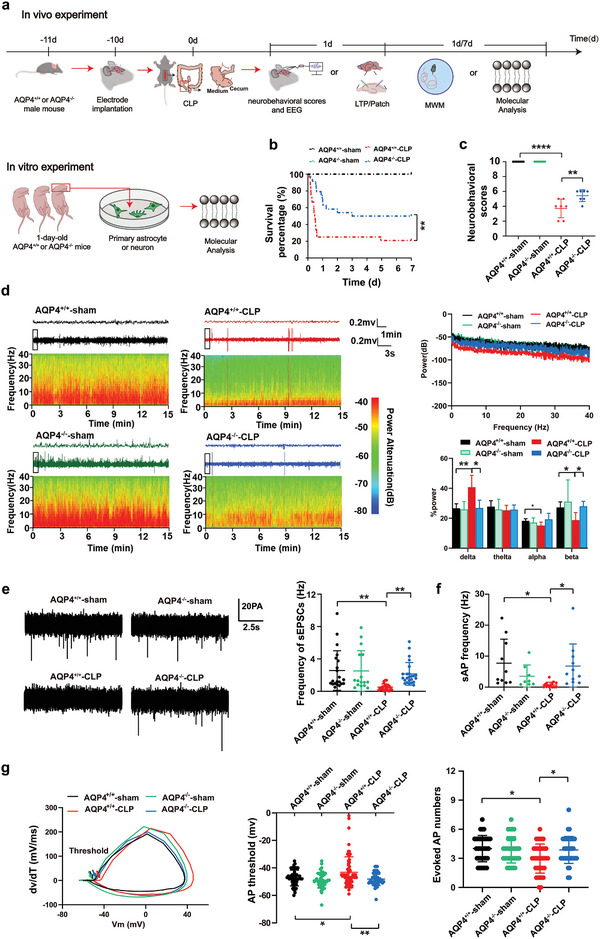
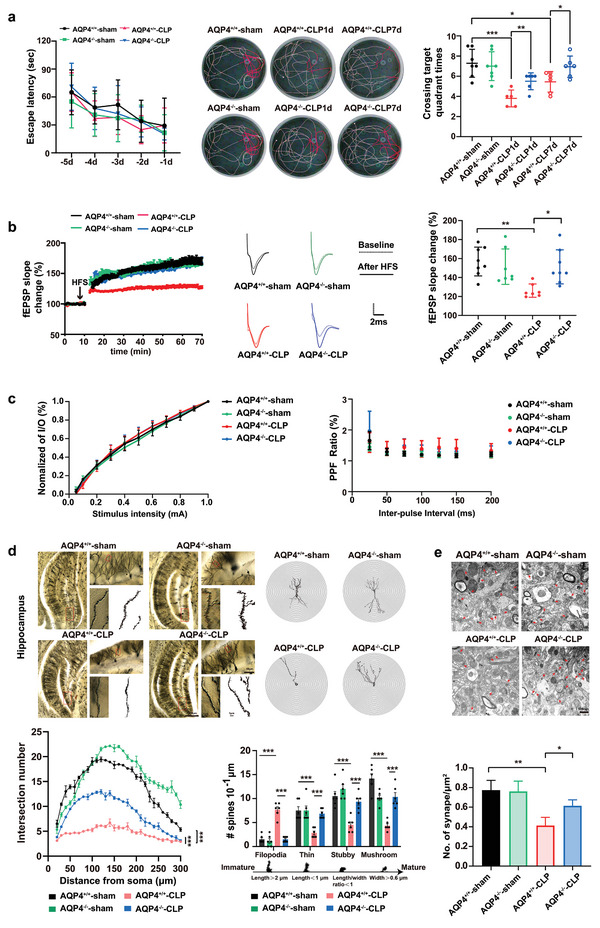
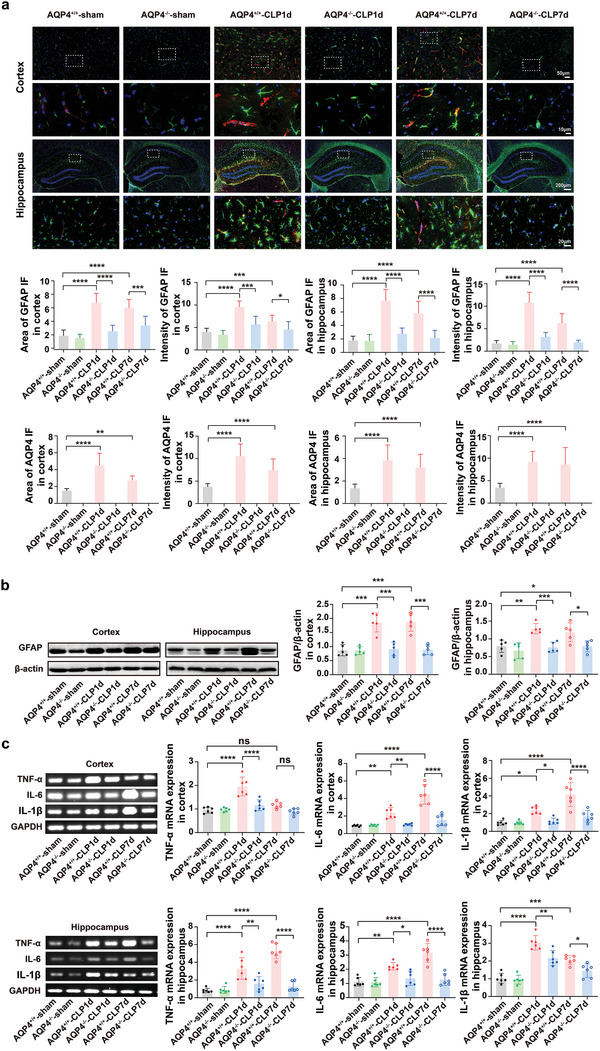
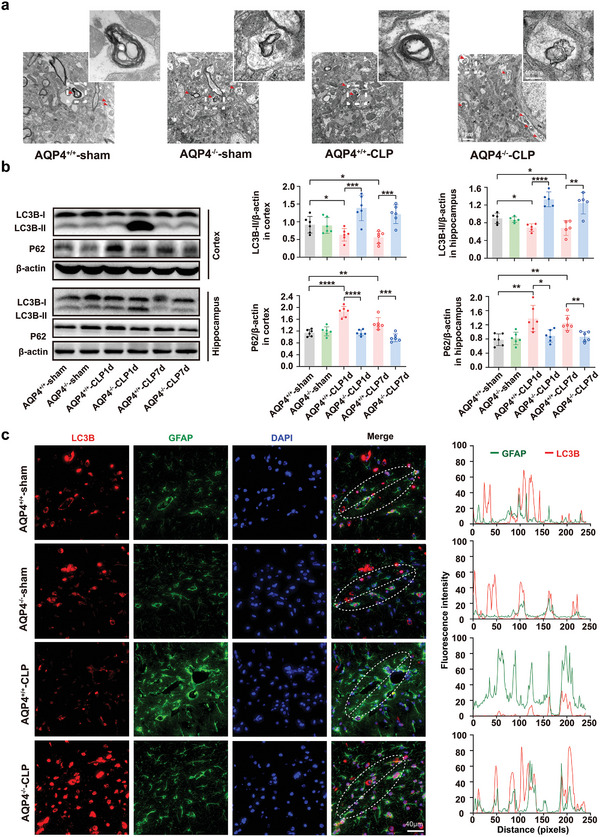
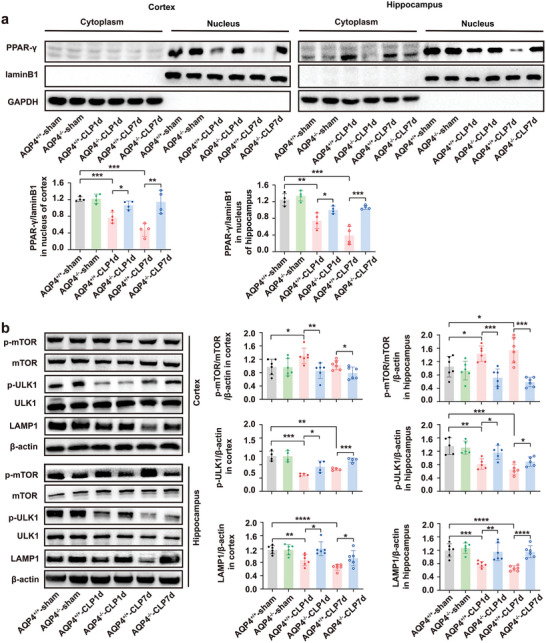
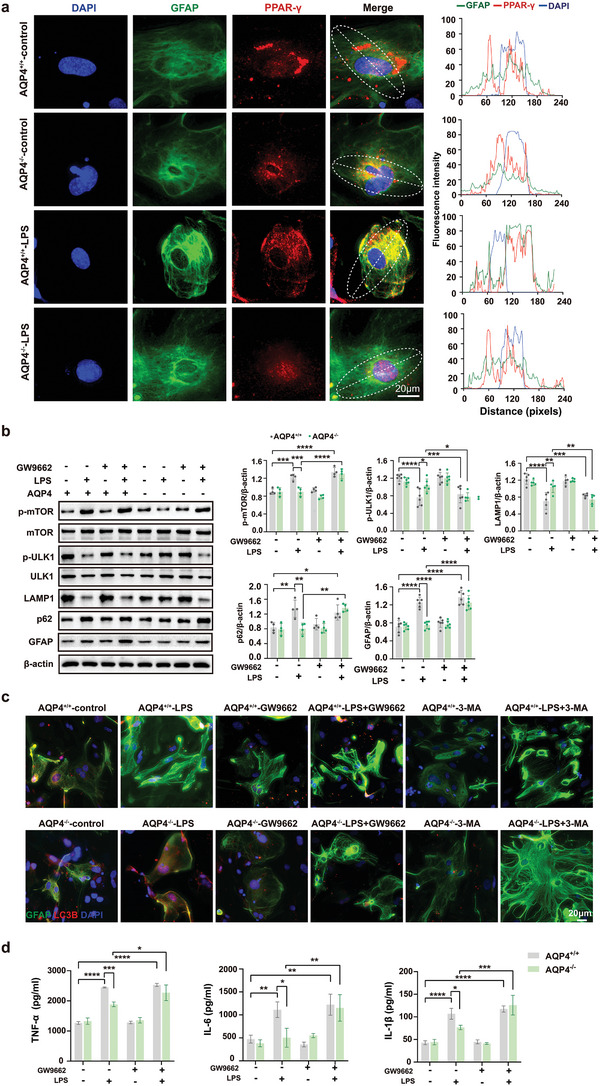
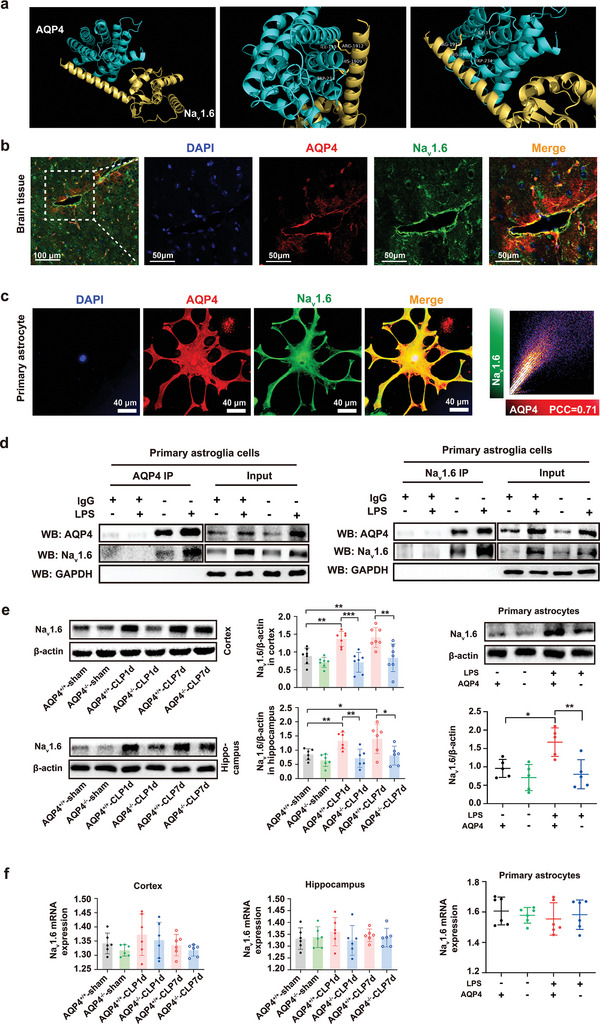
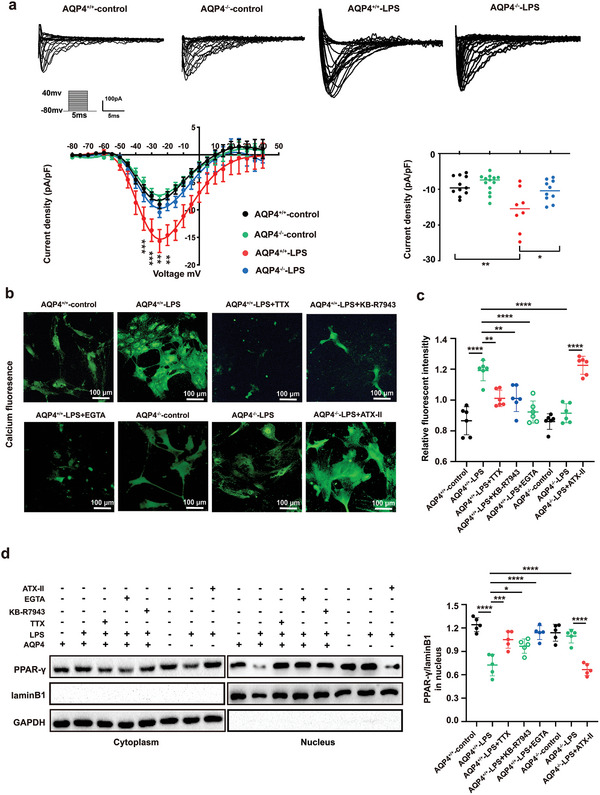
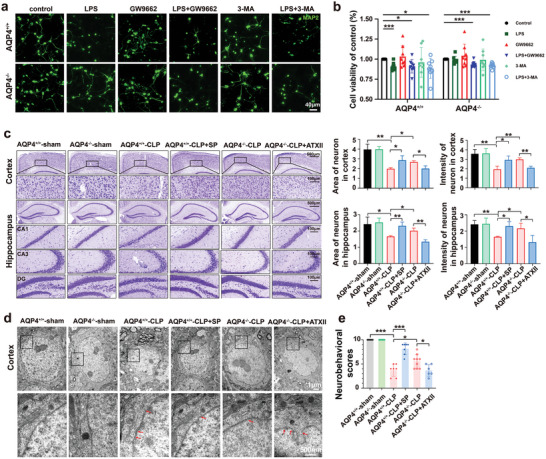
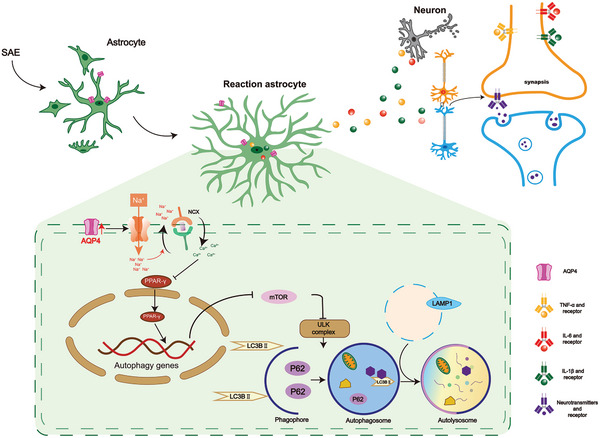
The pathology of sepsis-associated encephalopathy (SAE) is related to astrocyte-inflammation associated with aquaporin-4 (AQP4). The aim here is to investigate the effects of AQP4 associated with SAE and reveal its underlying mechanism causing cognitive impairment. The in vivo experimental results reveal that AQP4 in peripheral blood of patients with SAE is up-regulated, also the cortical and hippocampal tissue of cecal ligation and perforation (CLP) mouse brain has significant rise in AQP4. Furthermore, the data suggest that AQP4 deletion could attenuate learning and memory impairment, attributing to activation of astrocytic autophagy, inactivation of astrocyte and downregulate the expression of proinflammatory cytokines induced by CLP or lipopolysaccharide (LPS). Furthermore, the activation effect of AQP4 knockout on CLP or LPS-induced PPAR-γ inhibiting in astrocyte is related to intracellular Ca2+ level and sodium channel activity. Learning and memory impairment in SAE mouse model are attenuated by AQP4 knockout through activating autophagy, inhibiting neuroinflammation leading to neuroprotection via down-regulation of Nav 1.6 channels in the astrocytes. This results in the reduction of Ca2+ accumulation in the cell cytosol furthermore activating the inhibition of PPAR-γ signal transduction pathway in astrocytes.
Keywords: AQP4; astrocyte; autophagy; neuroinflammation; sepsis-associated encephalopathy; sodium channel Nav1.6.
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Thompson K., Venkatesh B., Finfer S., Intern. Med. J. 2019, 49, 160. - PubMed
-
- Pandharipande P. P., Girard T. D., Jackson J. C., Morandi A., Thompson J. L., Pun B. T., Brummel N. E., Hughes C. G., Vasilevskis E. E., Shintani A. K., Moons K. G., Geevarghese S. K., Canonico A., Hopkins R. O., Bernard G. R., Dittus R. S., Ely E. W., N. Engl. J. Med. 2013, 369, 1306. - PMC - PubMed
-
- Angus D. C., Linde‐Zwirble W. T., Lidicker J., Clermont G., Carcillo J., Pinsky M. R., Crit. Care Med. 2001, 29, 1303. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- LJKZZ20220097/Basic Scientific Research Project of Education Department of Liaoning Province
- LJKQZ2021101/Basic Scientific Research Project of Education Department of Liaoning Province
- 82073913/National Natural Sciences Foundation of China
- 82101275/National Natural Sciences Foundation of China
- 82101661/National Natural Sciences Foundation of China
- 81571061/National Natural Sciences Foundation of China
- 81671111/National Natural Sciences Foundation of China
- U1908208/National Natural Sciences Foundation of China
- 2021-MS-280/Natural Science Foundation of Liaoning Province
- 2022M713090/China Postdoctoral Science Foundation
- 2022LCYJYB06/The Second Hospital of Dalian Medical University 1+X program
LinkOut - more resources
Full Text Sources
Miscellaneous