

SHock-INduced Endotheliopathy (SHINE): A mechanistic justification for viscoelastography-guided resuscitation of traumatic and non-traumatic shock
- PMID: 36923287
- PMCID: PMC10009294
- DOI: 10.3389/fphys.2023.1094845
SHock-INduced Endotheliopathy (SHINE): A mechanistic justification for viscoelastography-guided resuscitation of traumatic and non-traumatic shock
Abstract
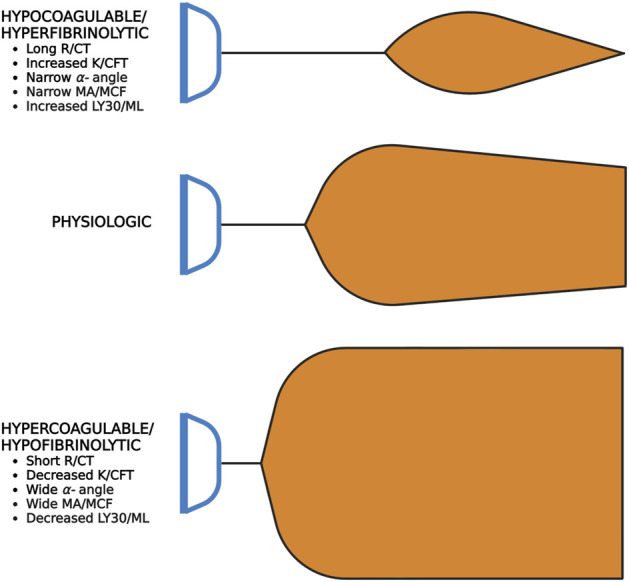
Irrespective of the reason for hypoperfusion, hypocoagulable and/or hyperfibrinolytic hemostatic aberrancies afflict up to one-quarter of critically ill patients in shock. Intensivists and traumatologists have embraced the concept of SHock-INduced Endotheliopathy (SHINE) as a foundational derangement in progressive shock wherein sympatho-adrenal activation may cause systemic endothelial injury. The pro-thrombotic endothelium lends to micro-thrombosis, enacting a cycle of worsening perfusion and increasing catecholamines, endothelial injury, de-endothelialization, and multiple organ failure. The hypocoagulable/hyperfibrinolytic hemostatic phenotype is thought to be driven by endothelial release of anti-thrombogenic mediators to the bloodstream and perivascular sympathetic nerve release of tissue plasminogen activator directly into the microvasculature. In the shock state, this hemostatic phenotype may be a counterbalancing, yet maladaptive, attempt to restore blood flow against a systemically pro-thrombotic endothelium and increased blood viscosity. We therefore review endothelial physiology with emphasis on glycocalyx function, unique biomarkers, and coagulofibrinolytic mediators, setting the stage for understanding the pathophysiology and hemostatic phenotypes of SHINE in various etiologies of shock. We propose that the hyperfibrinolytic phenotype is exemplified in progressive shock whether related to trauma-induced coagulopathy, sepsis-induced coagulopathy, or post-cardiac arrest syndrome-associated coagulopathy. Regardless of the initial insult, SHINE appears to be a catecholamine-driven entity which early in the disease course may manifest as hyper- or hypocoagulopathic and hyper- or hypofibrinolytic hemostatic imbalance. Moreover, these hemostatic derangements may rapidly evolve along the thrombohemorrhagic spectrum depending on the etiology, timing, and methods of resuscitation. Given the intricate hemochemical makeup and changes during these shock states, macroscopic whole blood tests of coagulative kinetics and clot strength serve as clinically useful and simple means for hemostasis phenotyping. We suggest that viscoelastic hemostatic assays such as thromboelastography (TEG) and rotational thromboelastometry (ROTEM) are currently the most applicable clinical tools for assaying global hemostatic function-including fibrinolysis-to enable dynamic resuscitation with blood products and hemostatic adjuncts for those patients with thrombotic and/or hemorrhagic complications in shock states.
Keywords: critical care; endothelium; glycocalyx; hemostasis; precision medicine; resuscitation; shock; thromboelastography.
Copyright © 2023 Bunch, Chang, Moore, Moore, Kwaan, Miller, Al-Fadhl, Thomas, Zackariya, Patel, Zackariya, Haidar, Patel, McCurdy, Thomas, Zimmer, Fulkerson, Kim, Walsh, Hake, Kedar, Aboukhaled and Walsh.
Conflict of interest statement
EM and HM have received research grants from Haemonetics Corporation outside the submitted work. Author MrW is employed by the company Cardinal Flow Assurance LLC. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer, AD, declared a shared parent affiliation with the author MM to the handling editor at the time of review.
Figures











References
-
- Adamzik M., Langemeier T., Frey U. H., Görlinger K., Saner F., Eggebrecht H., et al. (2011). Comparison of thrombelastometry with simplified acute physiology score II and sequential organ failure assessment scores for the prediction of 30-day survival: A cohort study. Shock 35 (4), 339–342. 10.1097/SHK.0b013e318204bff6 - DOI - PubMed
-
- Adrie C., Adib-Conquy M., Laurent I., Monchi M., Vinsonneau C., Fitting C., et al. (2002). Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation 106 (5), 562–568. 10.1161/01.cir.0000023891.80661.ad - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Medical