

Upregulation of acid ceramidase contributes to tumor progression in tuberous sclerosis complex
- PMID: 36927688
- PMCID: PMC10243802
- DOI: 10.1172/jci.insight.166850
Upregulation of acid ceramidase contributes to tumor progression in tuberous sclerosis complex
Abstract
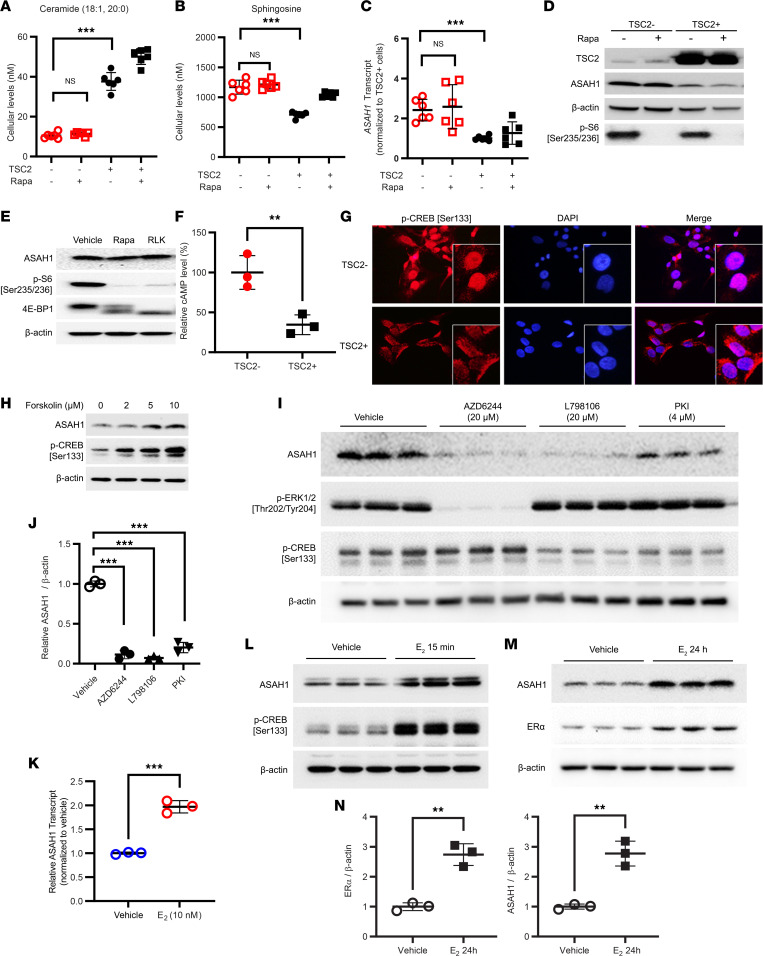
Tuberous sclerosis complex (TSC) is characterized by multisystem, low-grade neoplasia involving the lung, kidneys, brain, and heart. Lymphangioleiomyomatosis (LAM) is a progressive pulmonary disease affecting almost exclusively women. TSC and LAM are both caused by mutations in TSC1 and TSC2 that result in mTORC1 hyperactivation. Here, we report that single-cell RNA sequencing of LAM lungs identified activation of genes in the sphingolipid biosynthesis pathway. Accordingly, the expression of acid ceramidase (ASAH1) and dihydroceramide desaturase (DEGS1), key enzymes controlling sphingolipid and ceramide metabolism, was significantly increased in TSC2-null cells. TSC2 negatively regulated the biosynthesis of tumorigenic sphingolipids, and suppression of ASAH1 by shRNA or the inhibitor ARN14976 (17a) resulted in markedly decreased TSC2-null cell viability. In vivo, 17a significantly decreased the growth of TSC2-null cell-derived mouse xenografts and short-term lung colonization by TSC2-null cells. Combined rapamycin and 17a treatment synergistically inhibited renal cystadenoma growth in Tsc2+/- mice, consistent with increased ASAH1 expression and activity being rapamycin insensitive. Collectively, the present study identifies rapamycin-insensitive ASAH1 upregulation in TSC2-null cells and tumors and provides evidence that targeting aberrant sphingolipid biosynthesis pathways has potential therapeutic value in mechanistic target of rapamycin complex 1-hyperactive neoplasms, including TSC and LAM.
Keywords: Cell Biology; Metabolism; Molecular biology; Mouse models; Tumor suppressors.
Figures






References
-
- McCormack FX, Gupta N. Sporadic Lymphangioleiomyomatosis: Clinical Presentation and Diagnostic Evaluation. https://www.uptodate.com/contents/sporadic-lymphangioleiomyomatosis-clin... Updated: December 21, 2022. Accessed March 16, 2023.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
