Hyperactive nanobacteria with host-dependent traits pervade Omnitrophota
- PMID: 36928026
- PMCID: PMC10066038
- DOI: 10.1038/s41564-022-01319-1
Hyperactive nanobacteria with host-dependent traits pervade Omnitrophota
Abstract
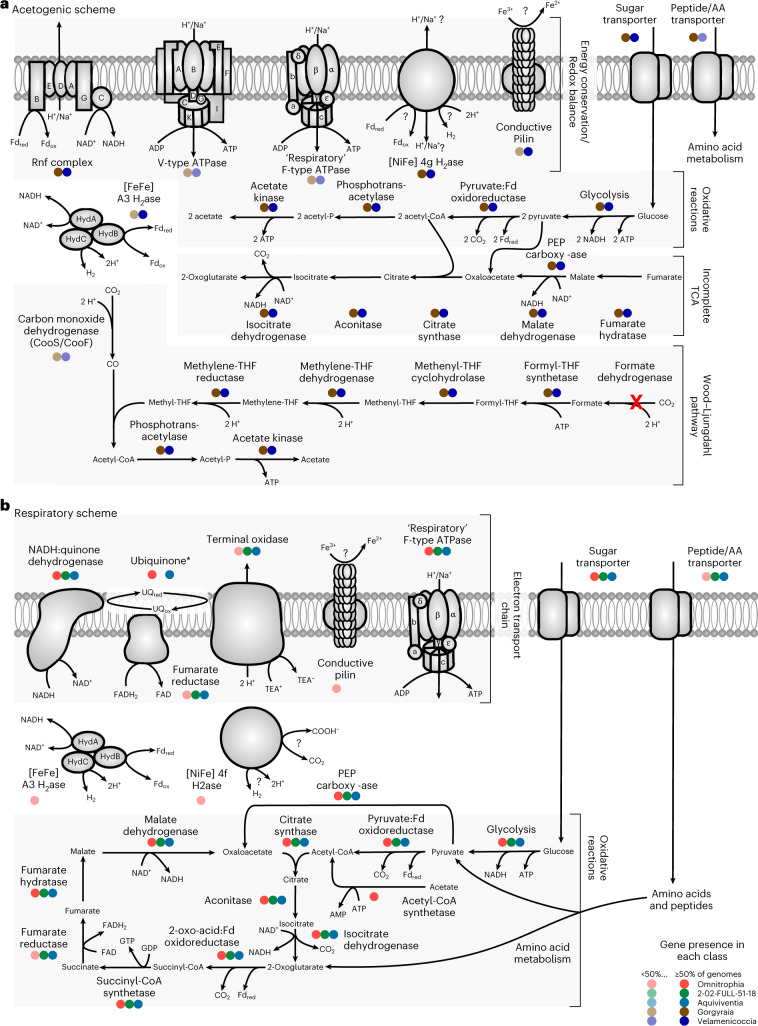
Candidate bacterial phylum Omnitrophota has not been isolated and is poorly understood. We analysed 72 newly sequenced and 349 existing Omnitrophota genomes representing 6 classes and 276 species, along with Earth Microbiome Project data to evaluate habitat, metabolic traits and lifestyles. We applied fluorescence-activated cell sorting and differential size filtration, and showed that most Omnitrophota are ultra-small (~0.2 μm) cells that are found in water, sediments and soils. Omnitrophota genomes in 6 classes are reduced, but maintain major biosynthetic and energy conservation pathways, including acetogenesis (with or without the Wood-Ljungdahl pathway) and diverse respirations. At least 64% of Omnitrophota genomes encode gene clusters typical of bacterial symbionts, suggesting host-associated lifestyles. We repurposed quantitative stable-isotope probing data from soils dominated by andesite, basalt or granite weathering and identified 3 families with high isotope uptake consistent with obligate bacterial predators. We propose that most Omnitrophota inhabit various ecosystems as predators or parasites.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures