

Kras oncogene ablation prevents resistance in advanced lung adenocarcinomas
- PMID: 36928090
- PMCID: PMC10065067
- DOI: 10.1172/JCI164413
Kras oncogene ablation prevents resistance in advanced lung adenocarcinomas
Abstract
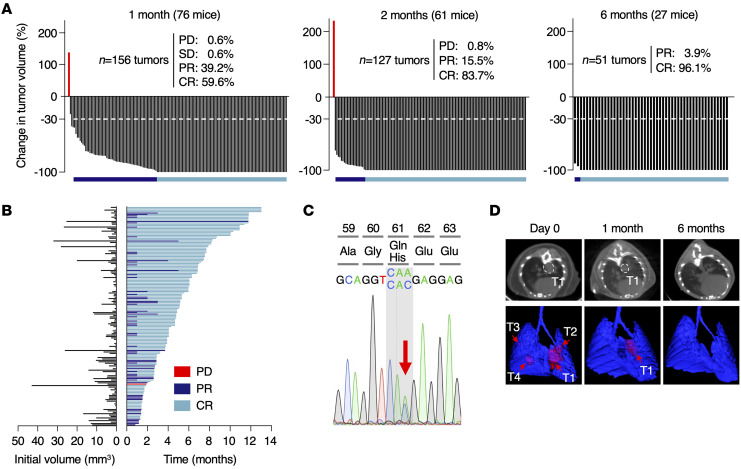
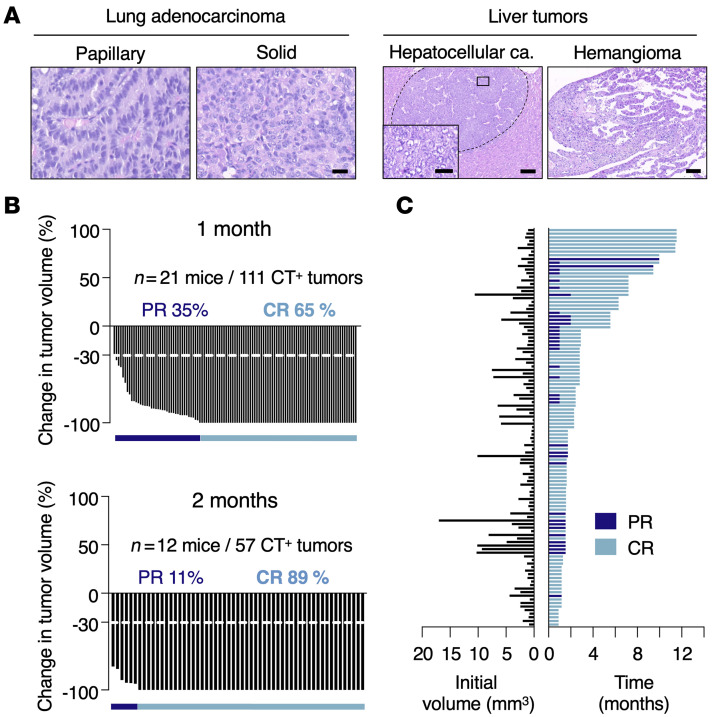
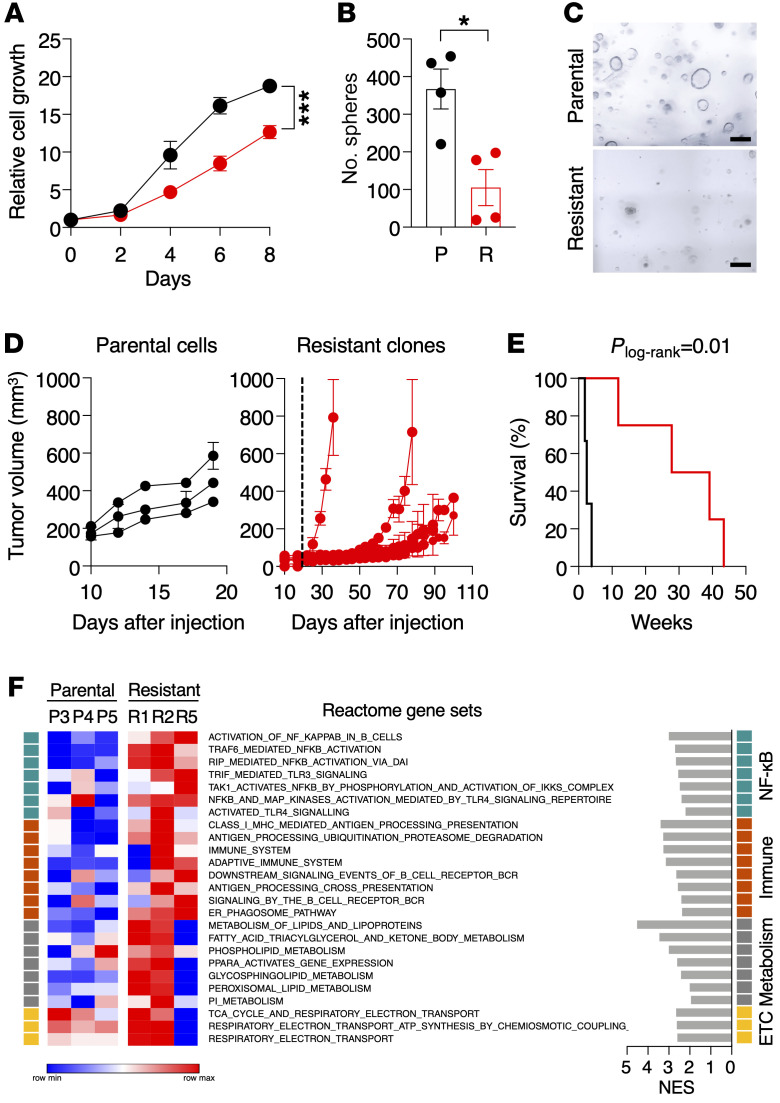
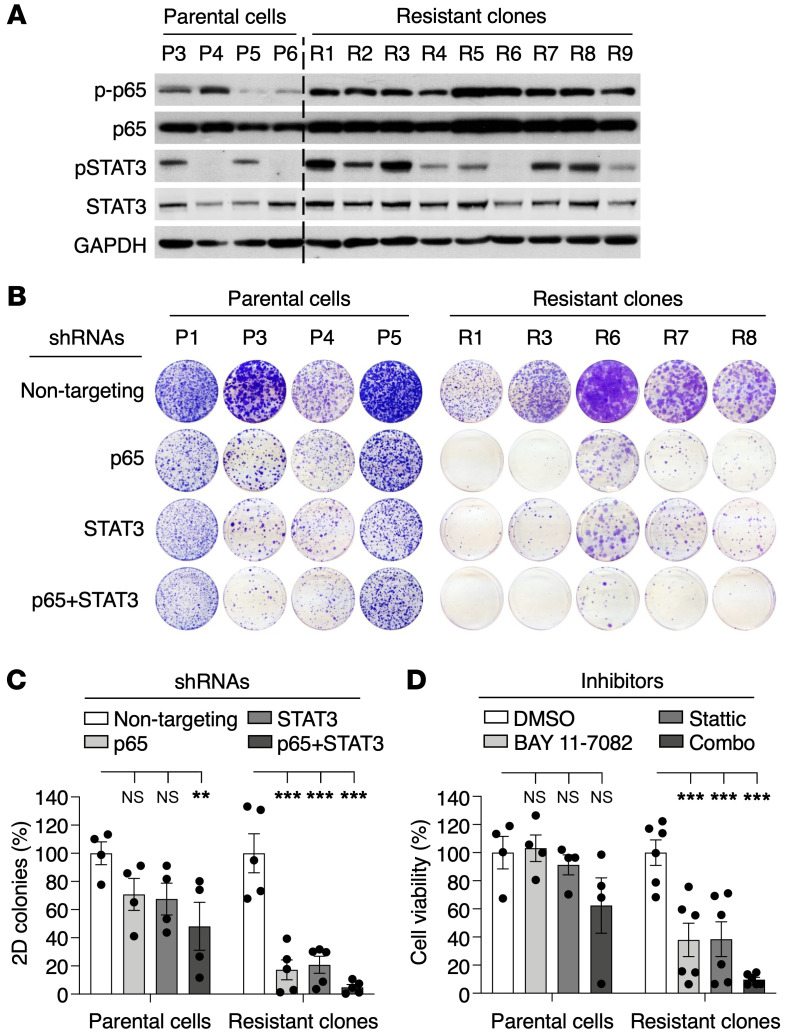
KRASG12C inhibitors have revolutionized the clinical management of patients with KRASG12C-mutant lung adenocarcinoma. However, patient exposure to these inhibitors leads to the rapid onset of resistance. In this study, we have used genetically engineered mice to compare the therapeutic efficacy and the emergence of tumor resistance between genetic ablation of mutant Kras expression and pharmacological inhibition of oncogenic KRAS activity. Whereas Kras ablation induces massive tumor regression and prevents the appearance of resistant cells in vivo, treatment of KrasG12C/Trp53-driven lung adenocarcinomas with sotorasib, a selective KRASG12C inhibitor, caused a limited antitumor response similar to that observed in the clinic, including the rapid onset of resistance. Unlike in human tumors, we did not observe mutations in components of the RAS-signaling pathways. Instead, sotorasib-resistant tumors displayed amplification of the mutant Kras allele and activation of xenobiotic metabolism pathways, suggesting that reduction of the on-target activity of KRASG12C inhibitors is the main mechanism responsible for the onset of resistance. In sum, our results suggest that resistance to KRAS inhibitors could be prevented by achieving a more robust inhibition of KRAS signaling mimicking the results obtained upon Kras ablation.
Keywords: Drug therapy; Lung cancer; Oncogenes; Oncology.
Figures
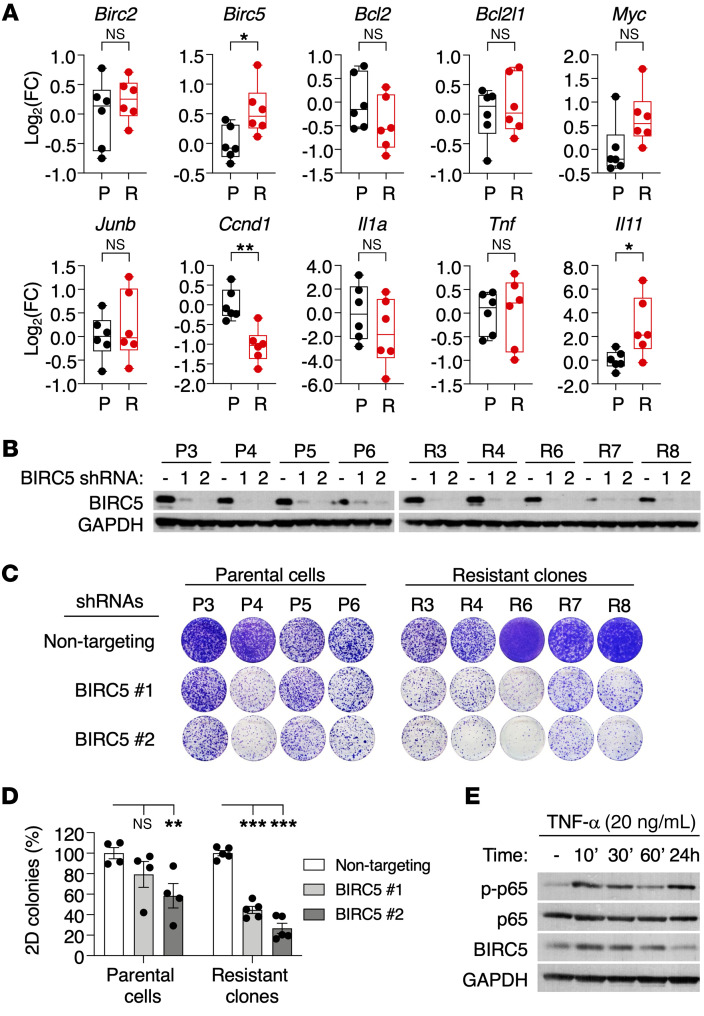
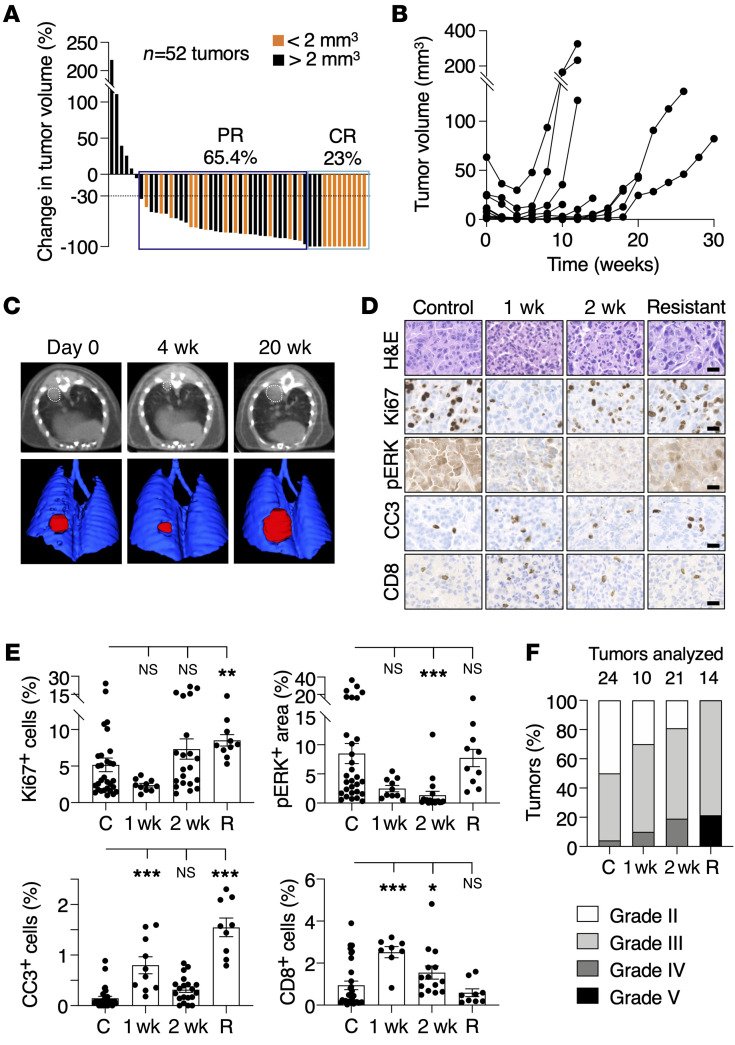
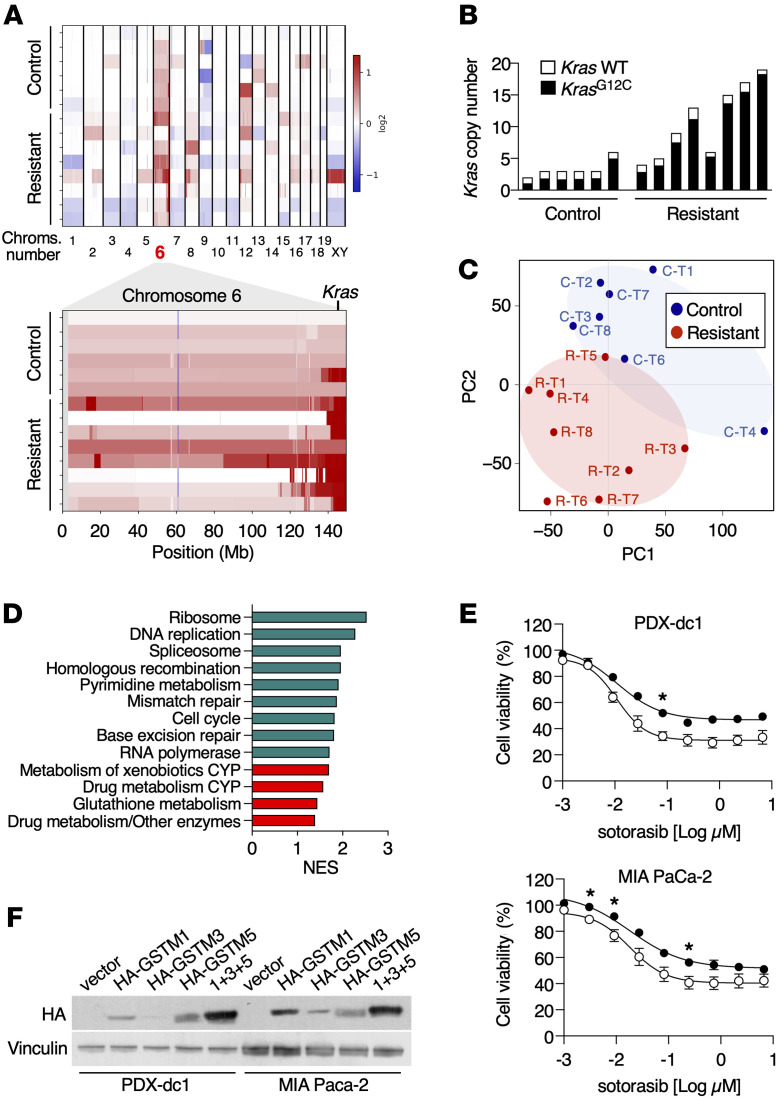
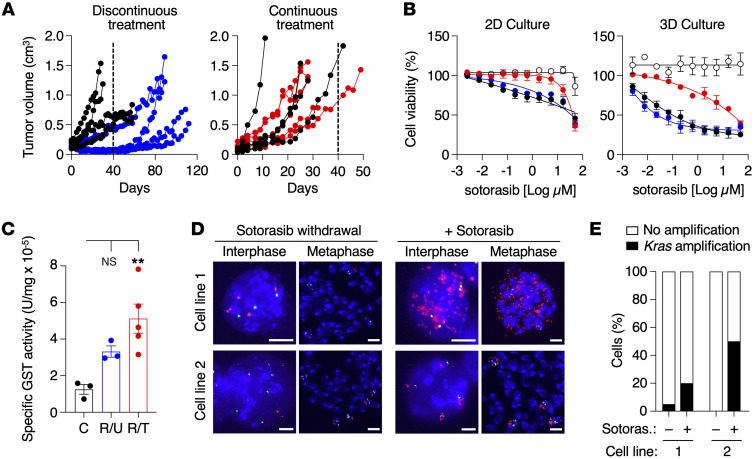
References
-
- Patricelli MP, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6(3):316–329. doi: 10.1158/2159-8290.CD-15-1105. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
