

Precise homology-directed installation of large genomic edits in human cells with cleaving and nicking high-specificity Cas9 variants
- PMID: 36928106
- PMCID: PMC10123109
- DOI: 10.1093/nar/gkad165
Precise homology-directed installation of large genomic edits in human cells with cleaving and nicking high-specificity Cas9 variants
Abstract
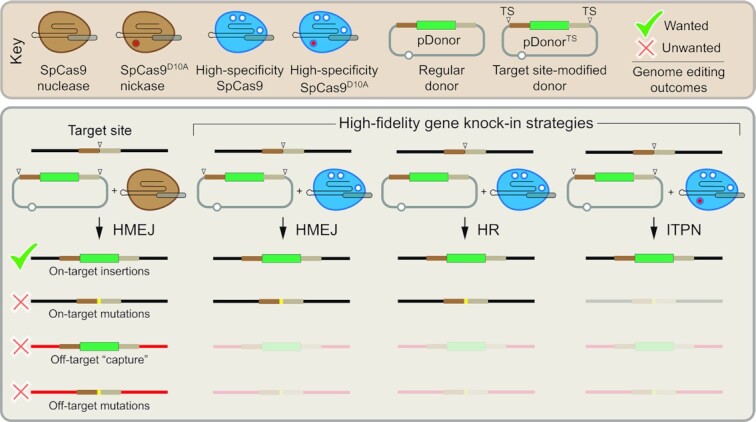
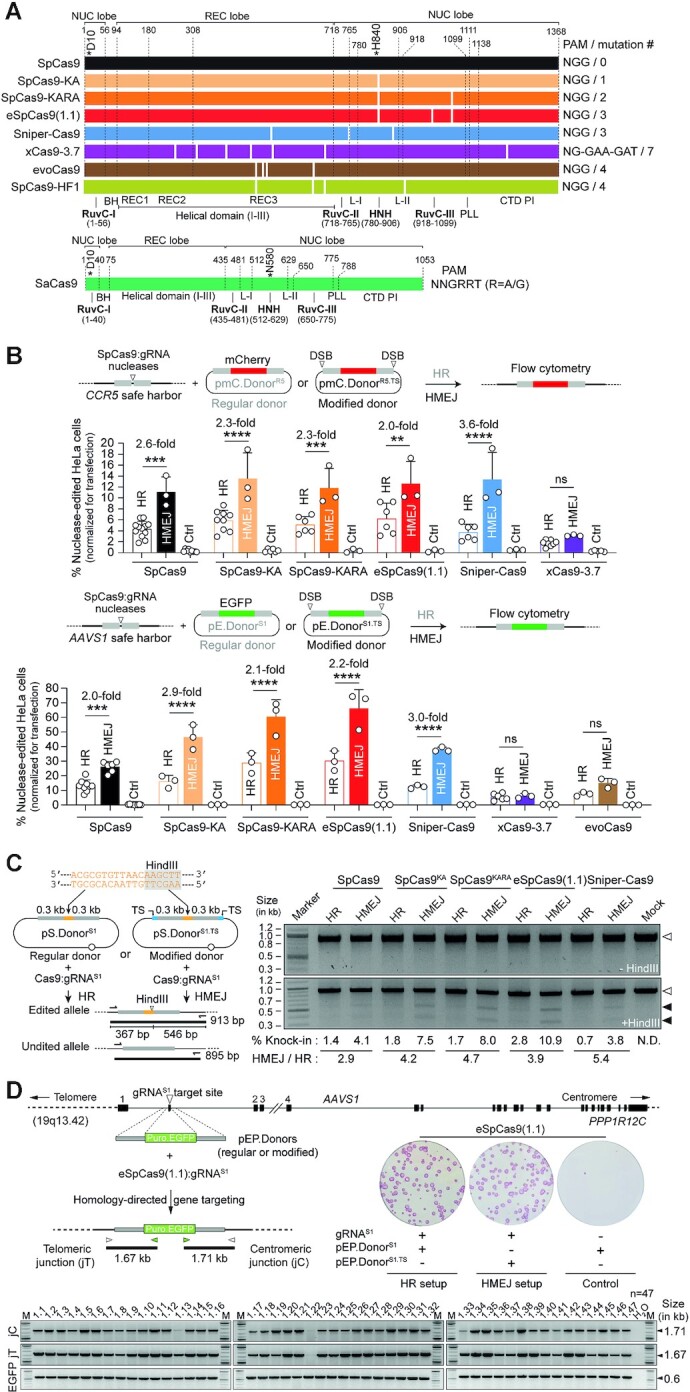
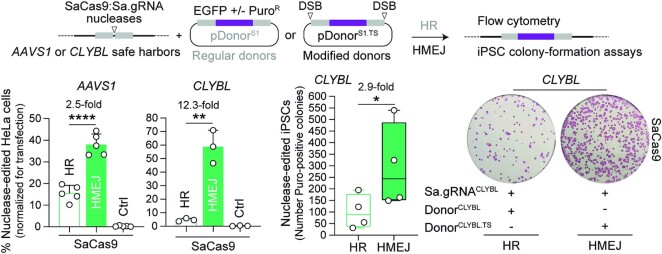
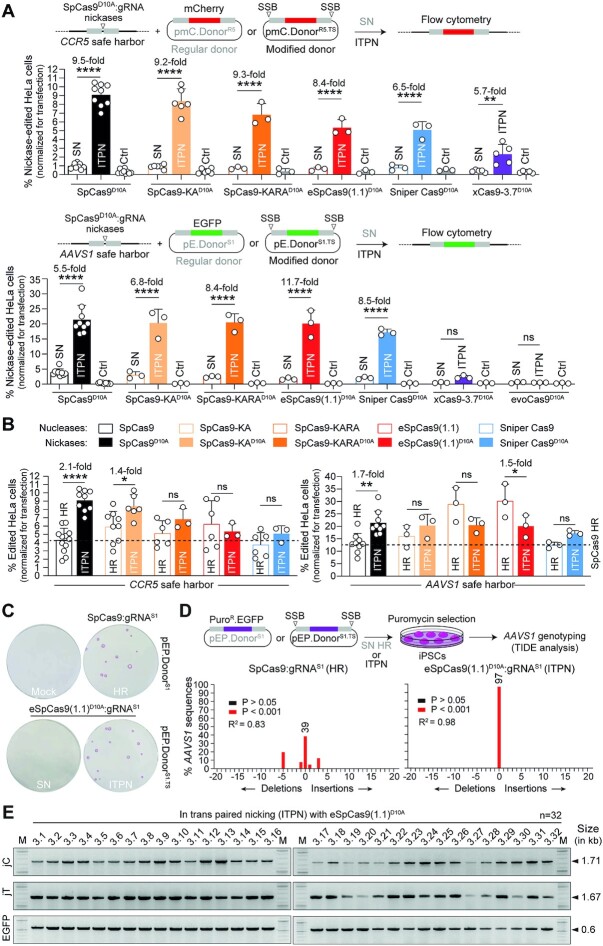
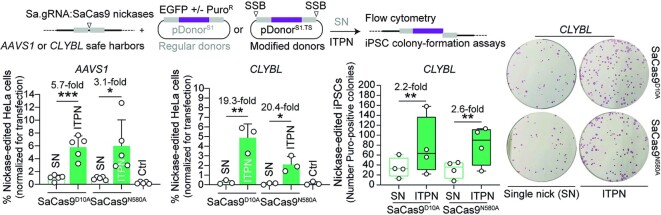
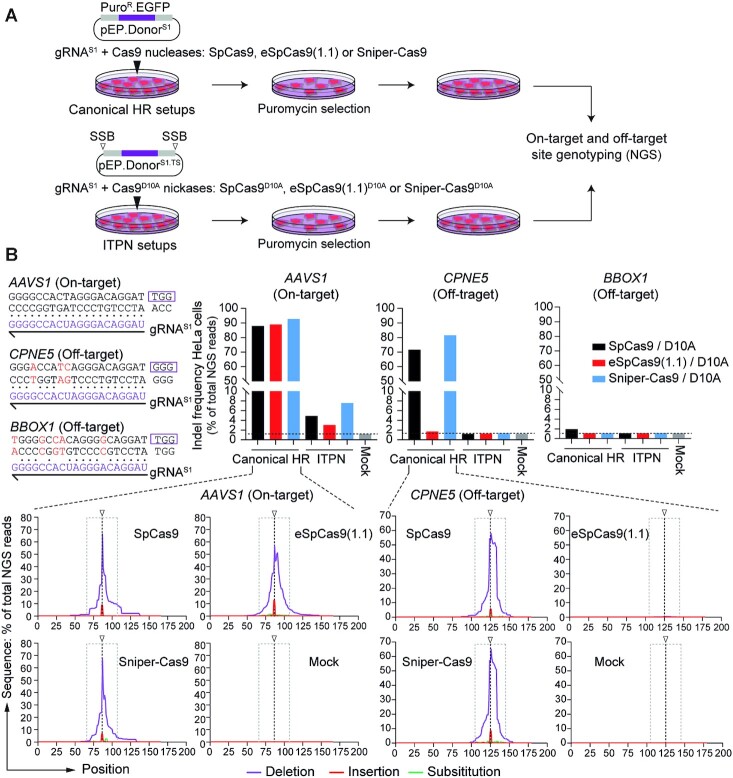
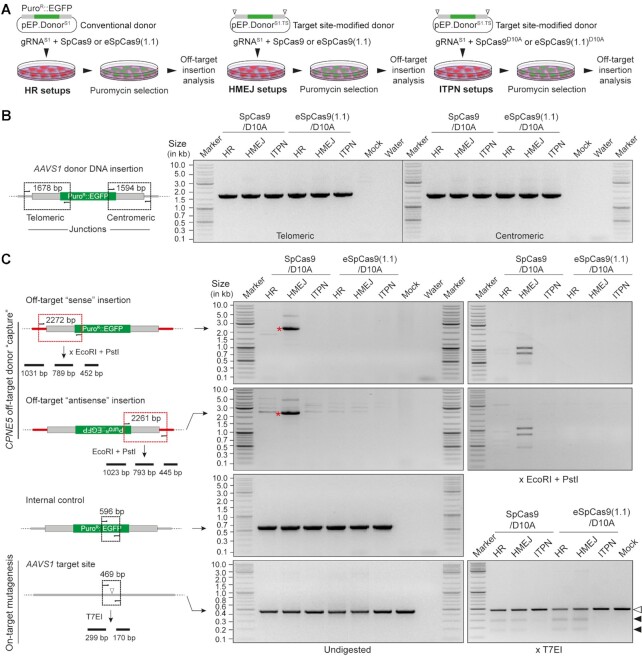
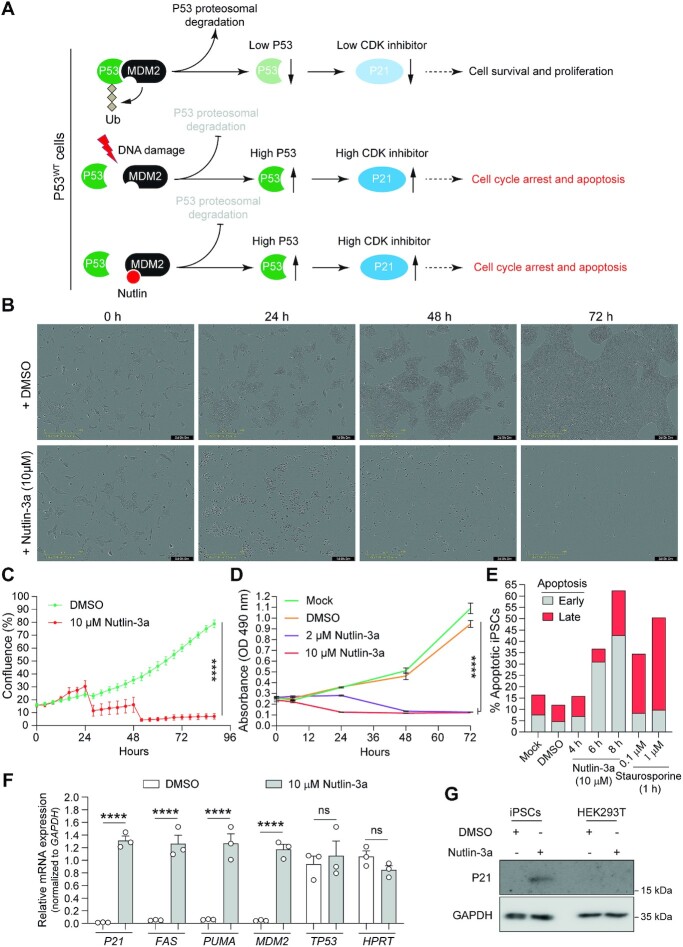
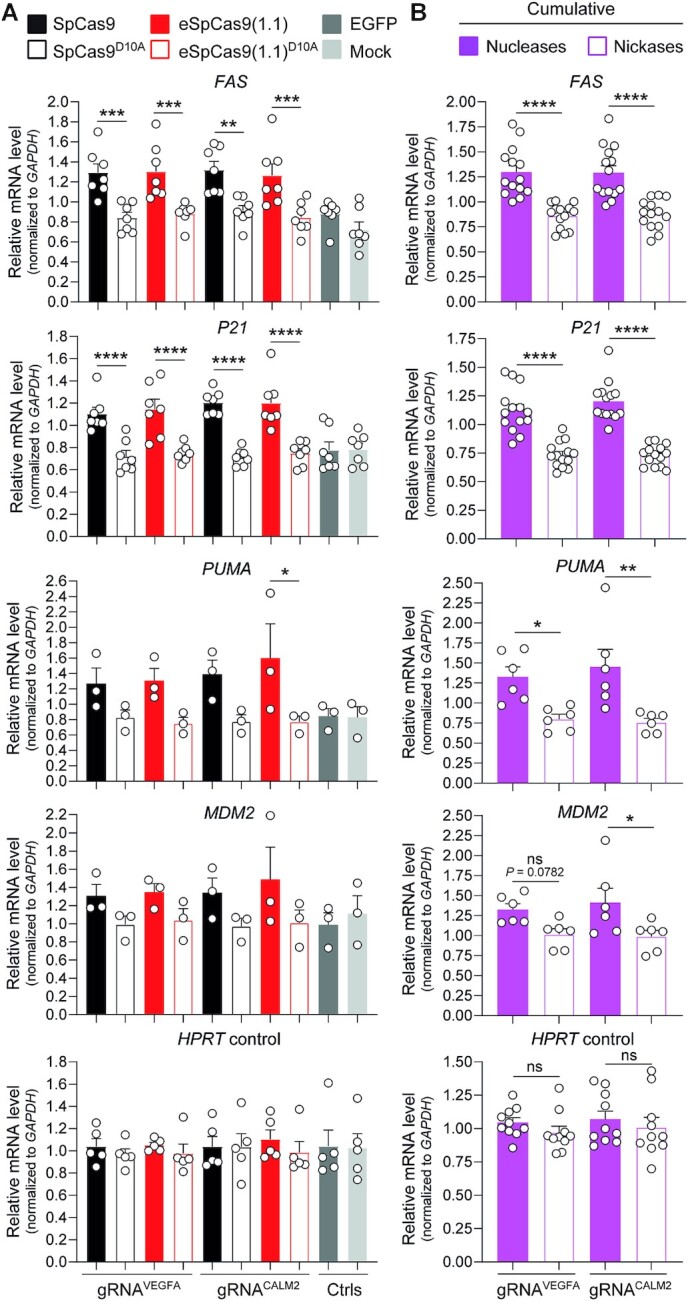
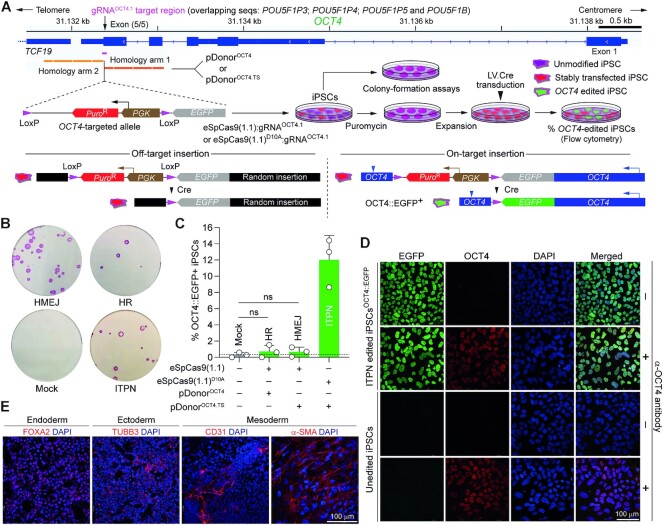
Homology-directed recombination (HDR) between donor constructs and acceptor genomic sequences cleaved by programmable nucleases, permits installing large genomic edits in mammalian cells in a precise fashion. Yet, next to precise gene knock-ins, programmable nucleases yield unintended genomic modifications resulting from non-homologous end-joining processes. Alternatively, in trans paired nicking (ITPN) involving tandem single-strand DNA breaks at target loci and exogenous donor constructs by CRISPR-Cas9 nickases, fosters seamless and scarless genome editing. In the present study, we identified high-specificity CRISPR-Cas9 nucleases capable of outperforming parental CRISPR-Cas9 nucleases in directing genome editing through homologous recombination (HR) and homology-mediated end joining (HMEJ) with donor constructs having regular and 'double-cut' designs, respectively. Additionally, we explored the ITPN principle by demonstrating its compatibility with orthogonal and high-specificity CRISPR-Cas9 nickases and, importantly, report that in human induced pluripotent stem cells (iPSCs), in contrast to high-specificity CRISPR-Cas9 nucleases, neither regular nor high-specificity CRISPR-Cas9 nickases activate P53 signaling, a DNA damage-sensing response linked to the emergence of gene-edited cells with tumor-associated mutations. Finally, experiments in human iPSCs revealed that differently from HR and HMEJ genome editing based on high-specificity CRISPR-Cas9 nucleases, ITPN involving high-specificity CRISPR-Cas9 nickases permits editing allelic sequences associated with essentiality and recurrence in the genome.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
