

Multiomic analysis of malignant pleural mesothelioma identifies molecular axes and specialized tumor profiles driving intertumor heterogeneity
- PMID: 36928603
- PMCID: PMC10101853
- DOI: 10.1038/s41588-023-01321-1
Multiomic analysis of malignant pleural mesothelioma identifies molecular axes and specialized tumor profiles driving intertumor heterogeneity
Abstract
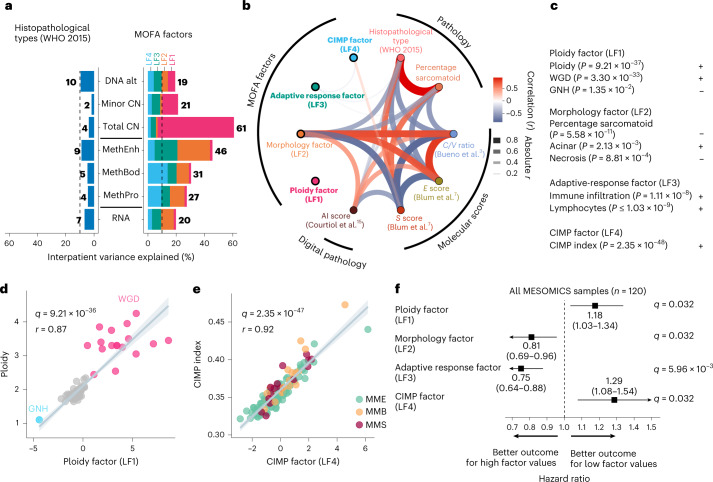
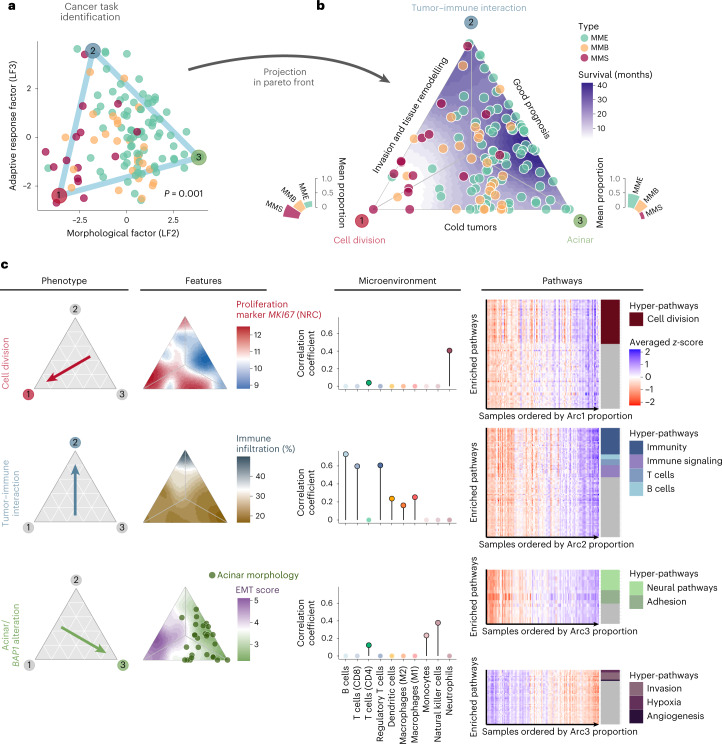
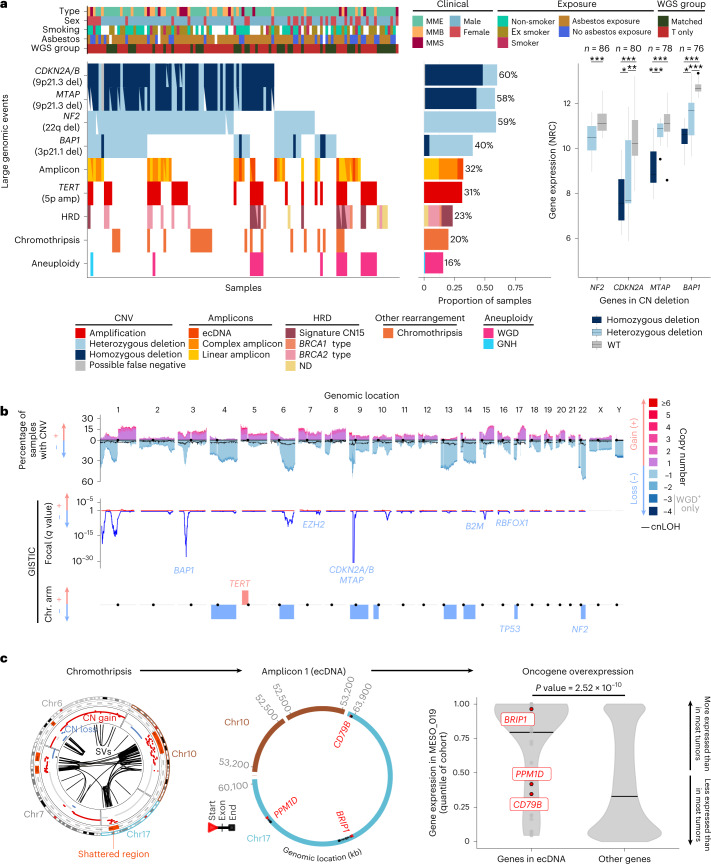
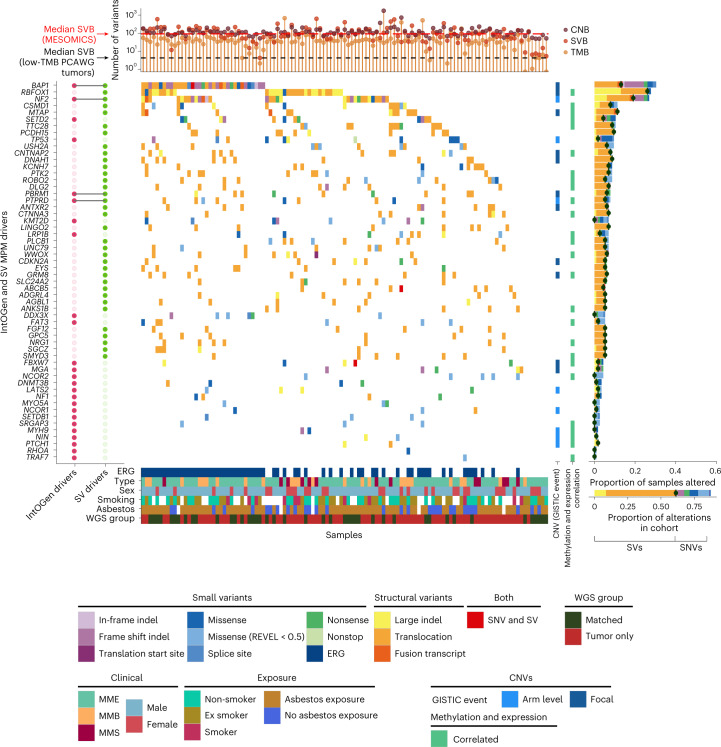
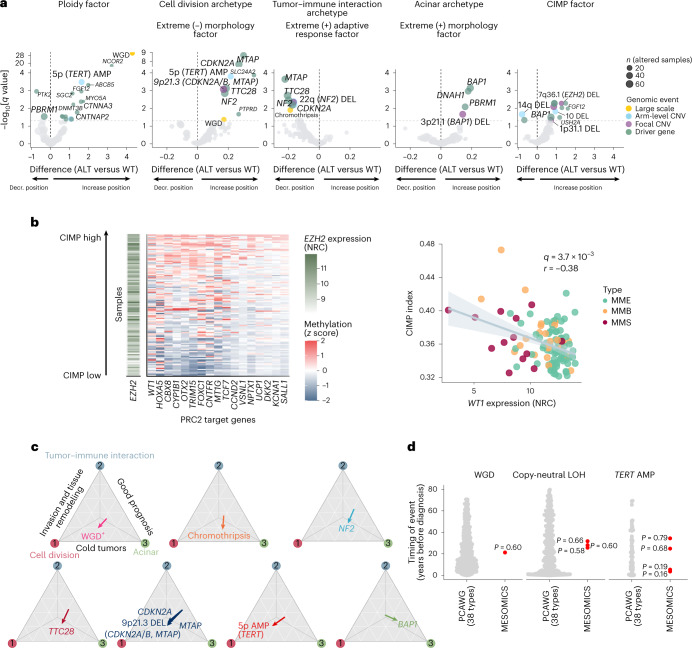
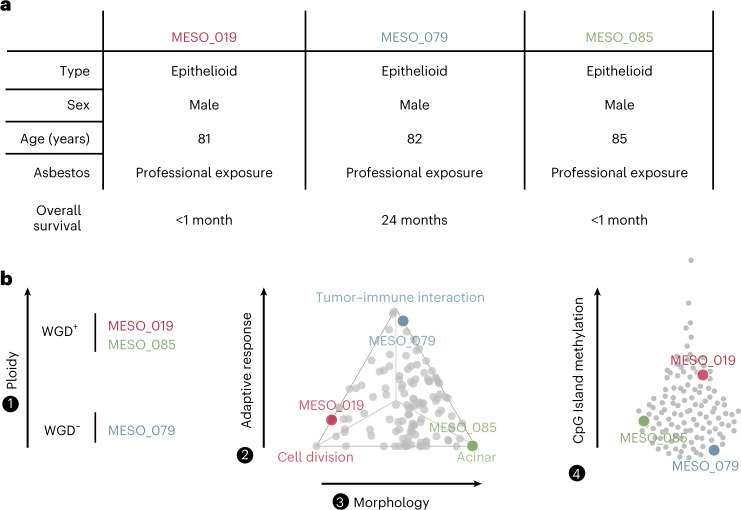
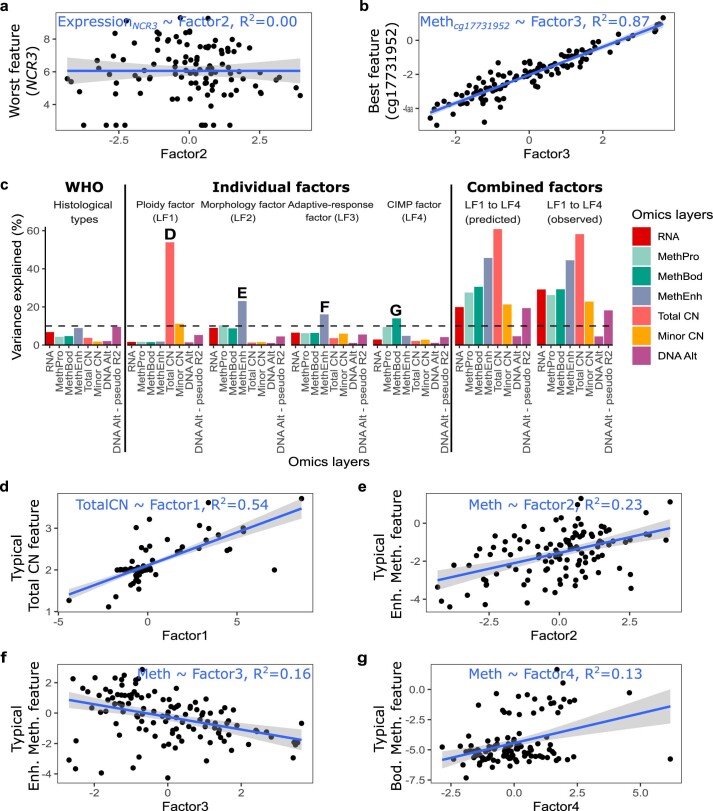
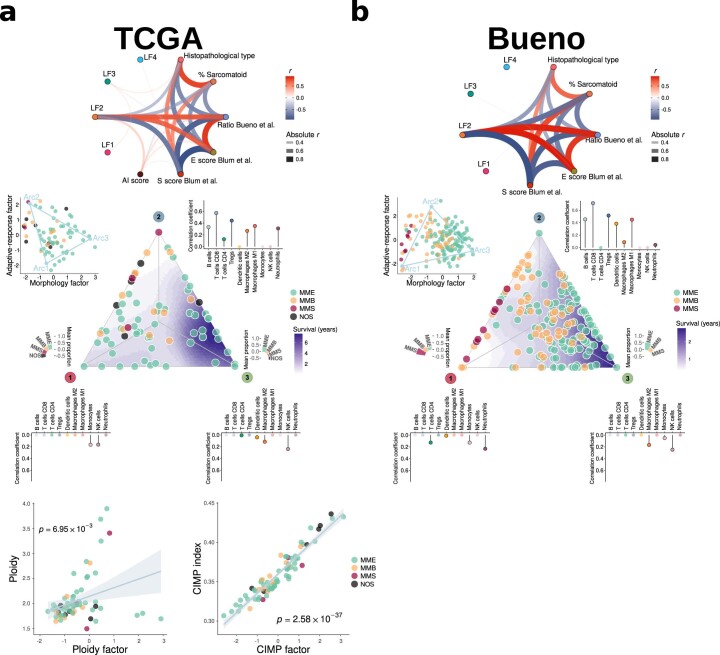
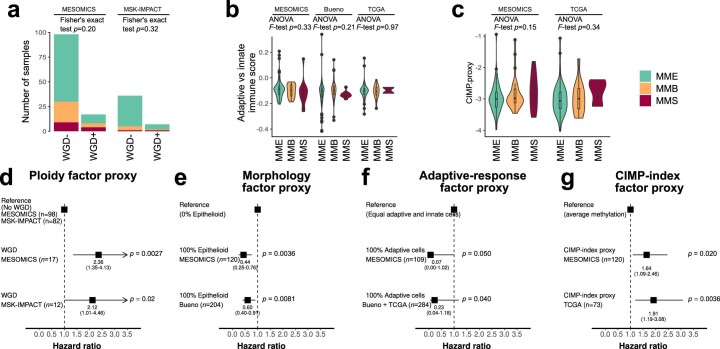
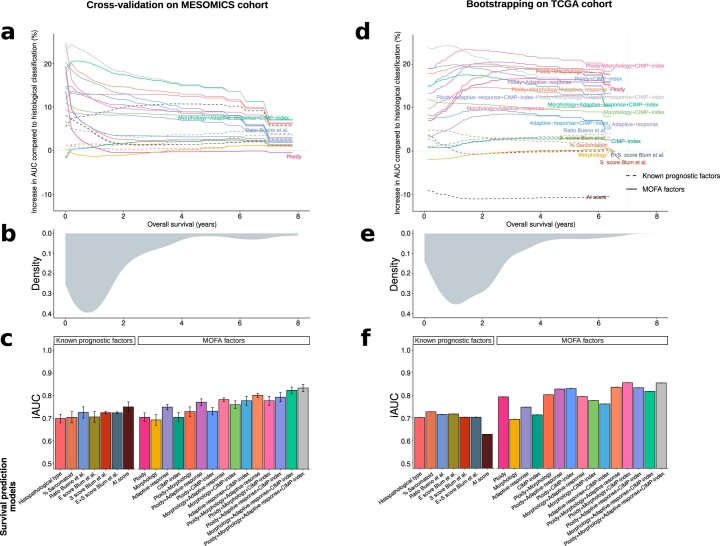
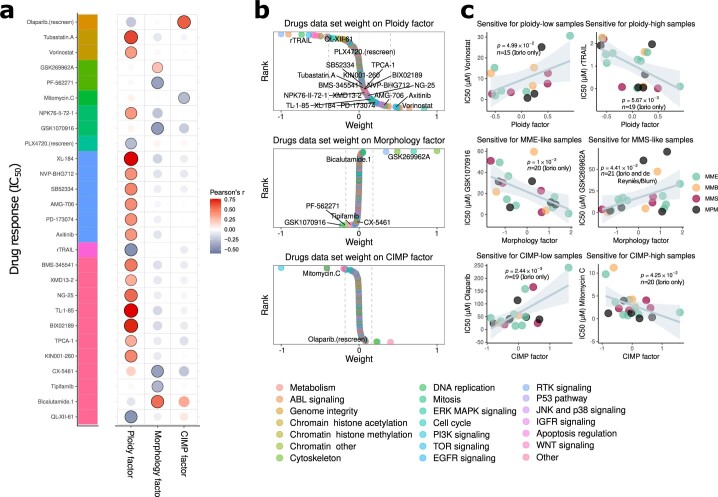
Malignant pleural mesothelioma (MPM) is an aggressive cancer with rising incidence and challenging clinical management. Through a large series of whole-genome sequencing data, integrated with transcriptomic and epigenomic data using multiomics factor analysis, we demonstrate that the current World Health Organization classification only accounts for up to 10% of interpatient molecular differences. Instead, the MESOMICS project paves the way for a morphomolecular classification of MPM based on four dimensions: ploidy, tumor cell morphology, adaptive immune response and CpG island methylator profile. We show that these four dimensions are complementary, capture major interpatient molecular differences and are delimited by extreme phenotypes that-in the case of the interdependent tumor cell morphology and adapted immune response-reflect tumor specialization. These findings unearth the interplay between MPM functional biology and its genomic history, and provide insights into the variations observed in the clinical behavior of patients with MPM.
© 2023. The Author(s).
Conflict of interest statement
Where authors are identified as personnel of the IARC/WHO, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the IARC/WHO. Where authors are identified as personnel of the Centre de Recherche en Cancérologie de Lyon, the authors declare no competing interests. A.S. participated in expert boards and clinical trials with AstraZeneca, Bristol-Myers Squibb, MSD and Roche. N.G. declares consultancy for and research support from Bristol-Myers Squibb, AstraZeneca, Roche and MSD. S Lantuejoul declares research support from AstraZeneca, Sanofi, Bristol-Myers Squibb, Janssen and Eli Lilly and has participated in expert boards for MSD and Bristol-Myers Squibb. D.D. declares research support from AstraZeneca. J. Mazieres declares consultancy for and research support from Roche, AstraZeneca, Bristol-Myers Squibb, MSD and Pierre Fabre. M.B. declares consultancy for and research support from AstraZeneca, Bristol-Myers Squibb and Amgen. I.R. participated in expert boards for AstraZeneca, MSD and Bristol-Myers Squibb. L.B.A. is a compensated consultant and has equity interest in io9. His spouse is an employee of Biotheranostics. L.B.A. is also an inventor on a US patent (10,776,718) relating to source identification by non-negative matrix factorization. E.N.B. and L.B.A. declare US provisional patent applications with the serial numbers 63/289,601 and 63/269,033. L.B.A. declares US provisional patent applications with the serial numbers 63/366,392, 63/367,846 and 63/412,835. C.M. is employed by and has equity interest in Owkin. C.M., P.C. and F.G.-S. are inventors on the US patent 17185924 ‘Systems and methods for mesothelioma feature detection and enhanced prognosis or response to treatment’. All other authors declare no competing interests.
Figures
References
-
- WHO Classification of Tumours, Thoracic Tumours (5th edn) (International Agency for Research on Cancer, 2020).
-
- Hmeljak J, et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018;8:1548–1565. doi: 10.1158/2159-8290.CD-18-0804. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
