

A longitudinal circulating tumor DNA-based model associated with survival in metastatic non-small-cell lung cancer
- PMID: 36928816
- PMCID: PMC10115641
- DOI: 10.1038/s41591-023-02226-6
A longitudinal circulating tumor DNA-based model associated with survival in metastatic non-small-cell lung cancer
Abstract
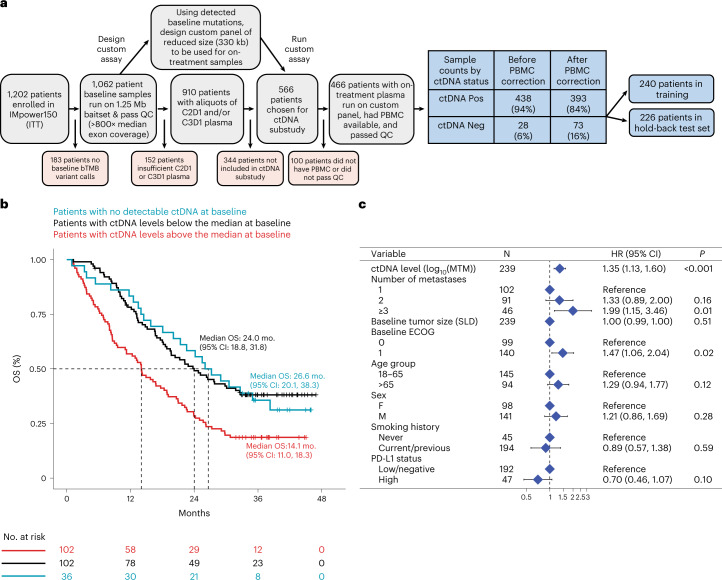
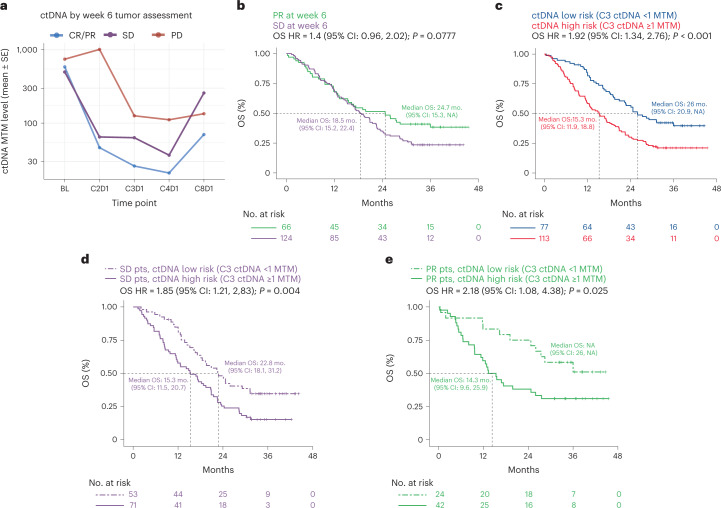
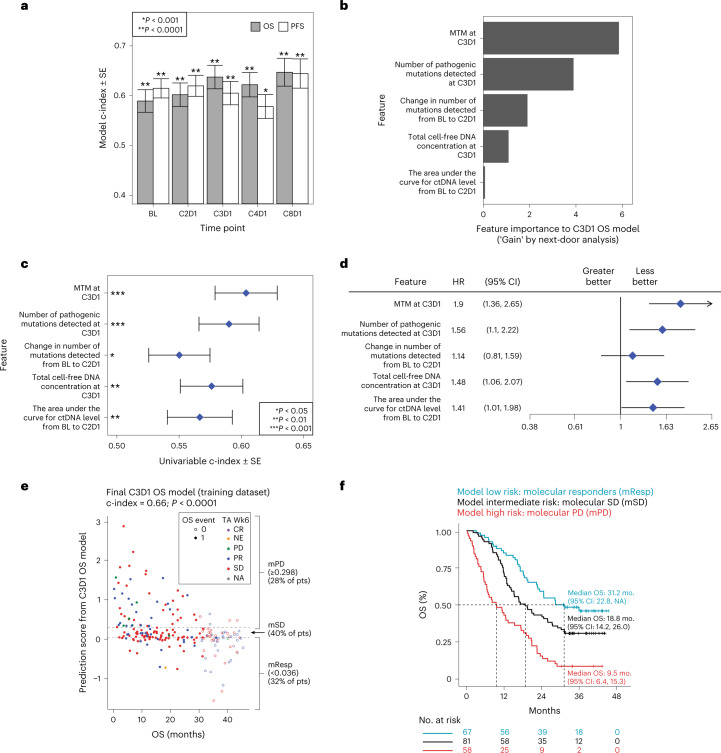
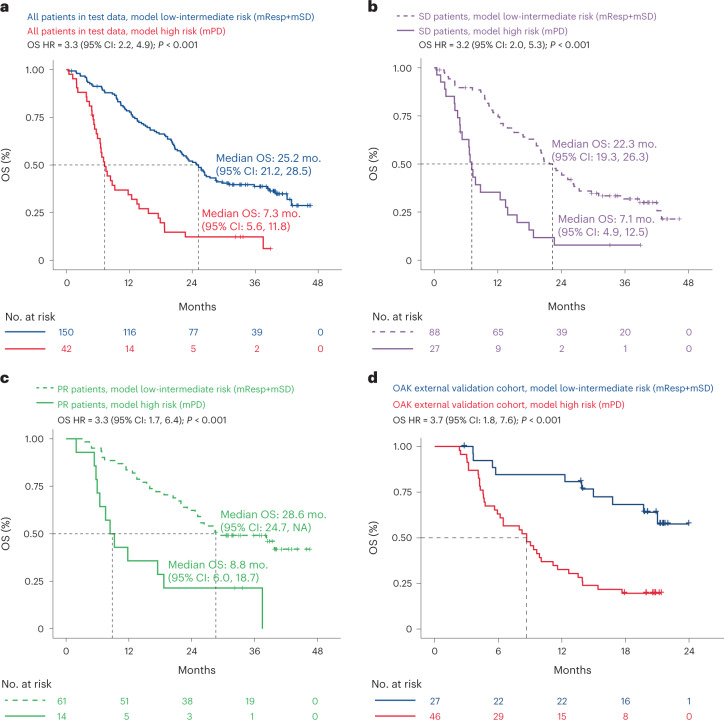
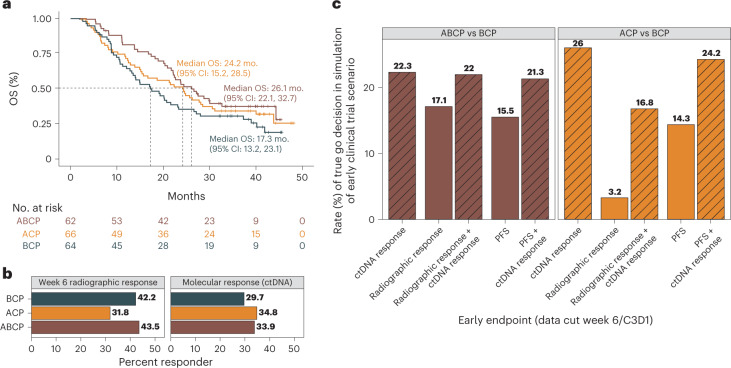
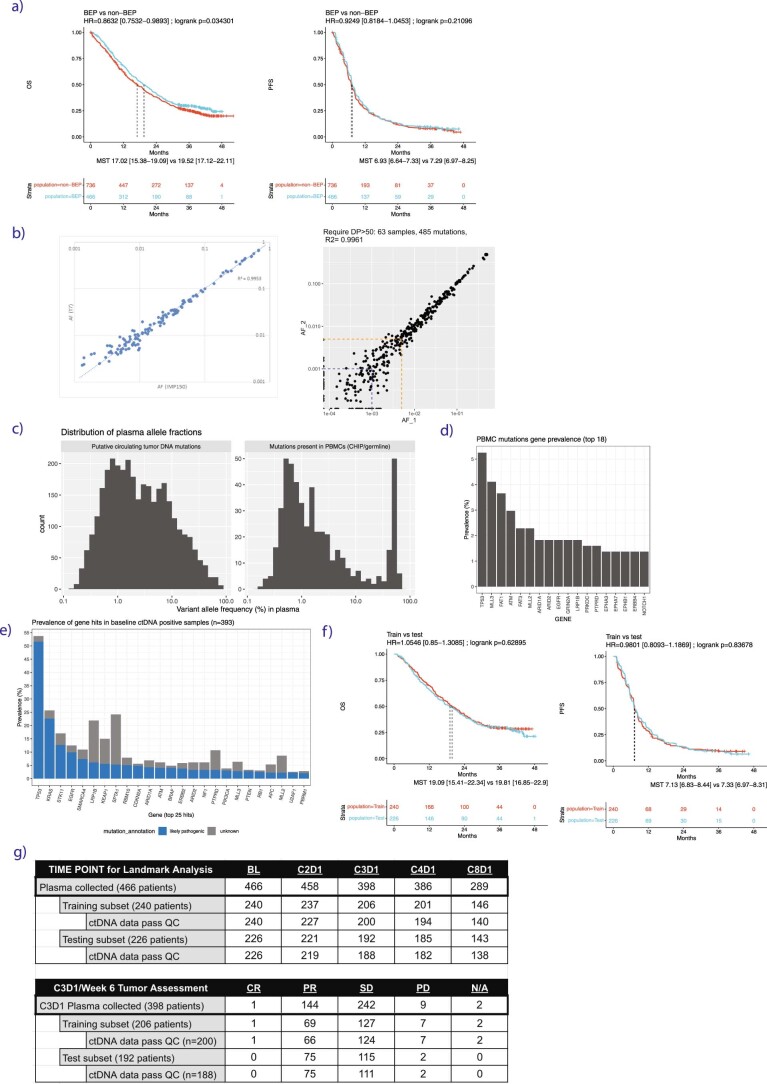
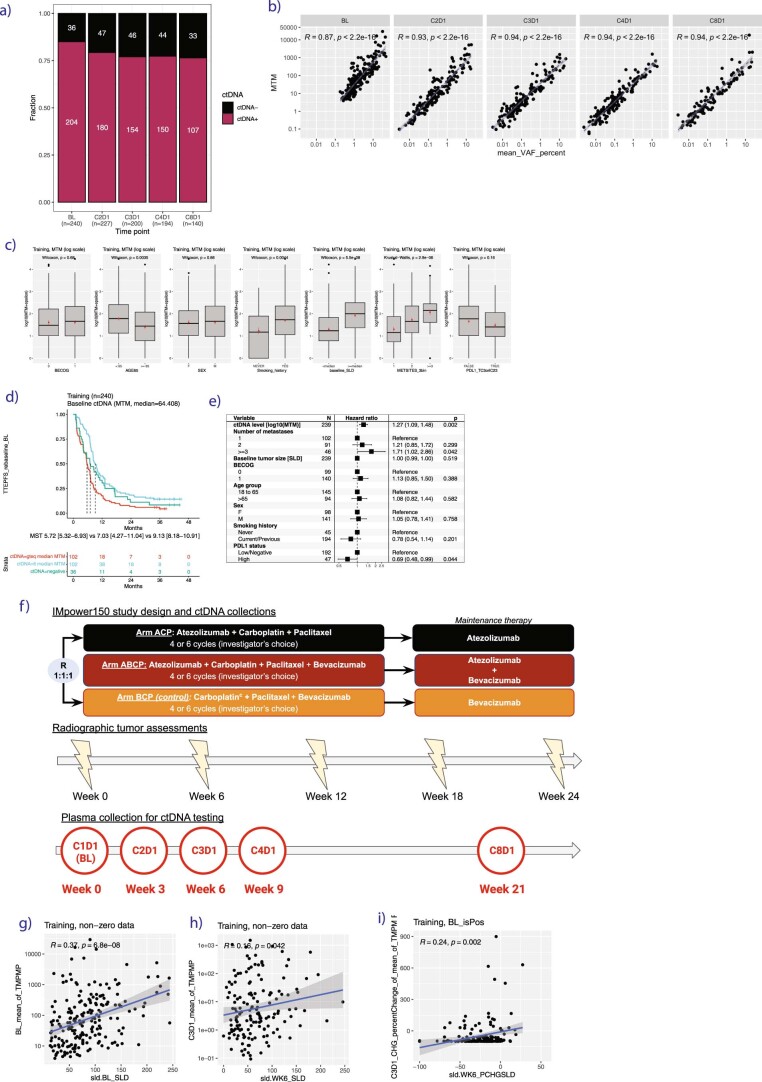
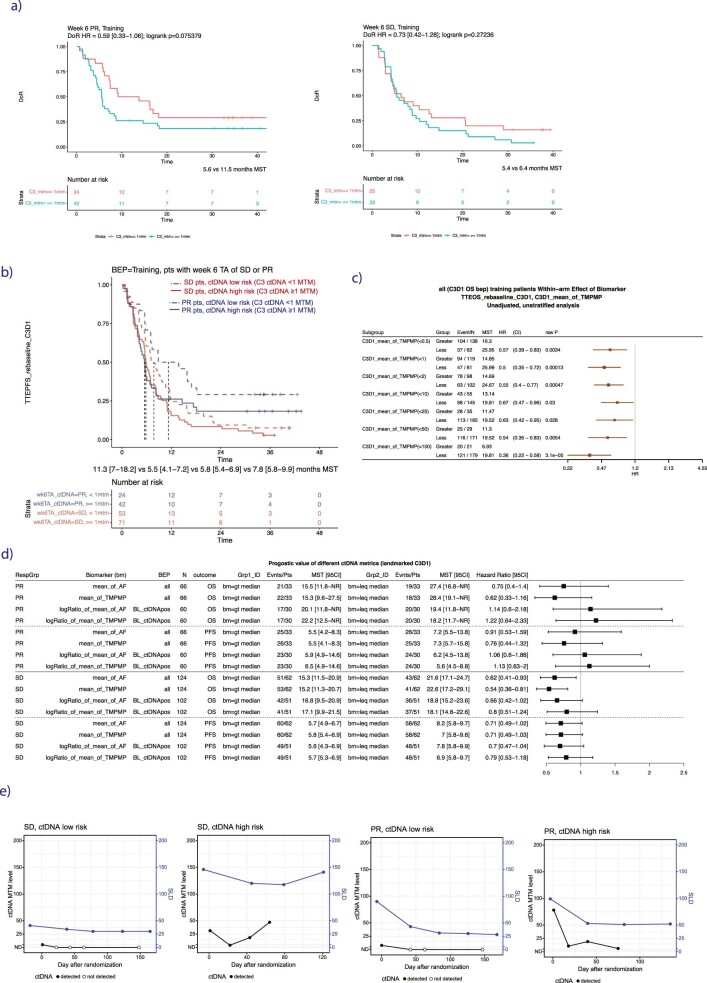
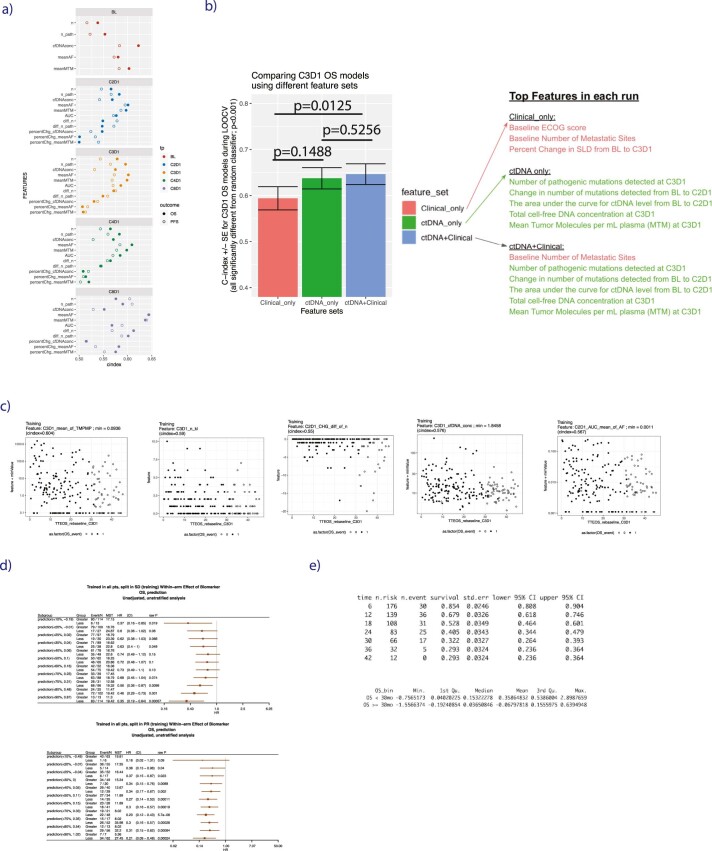
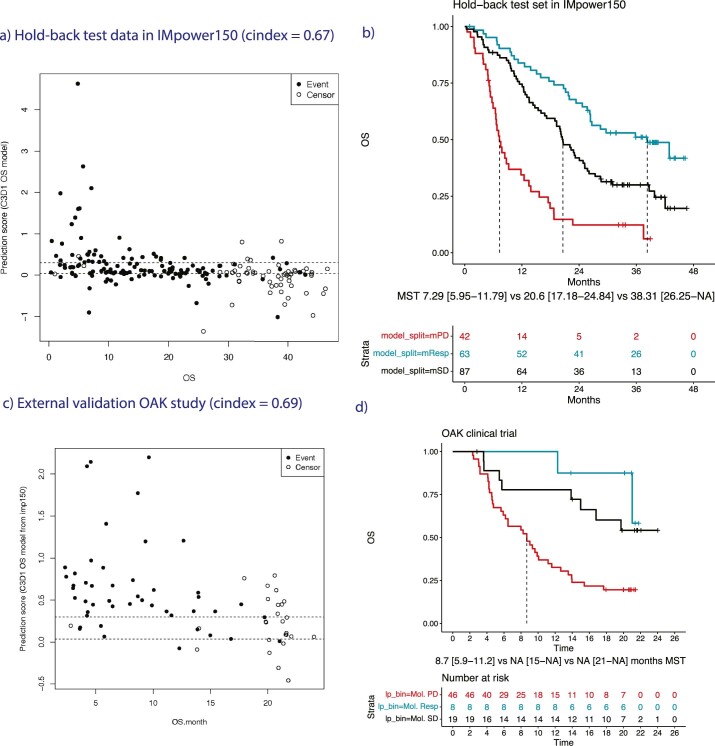
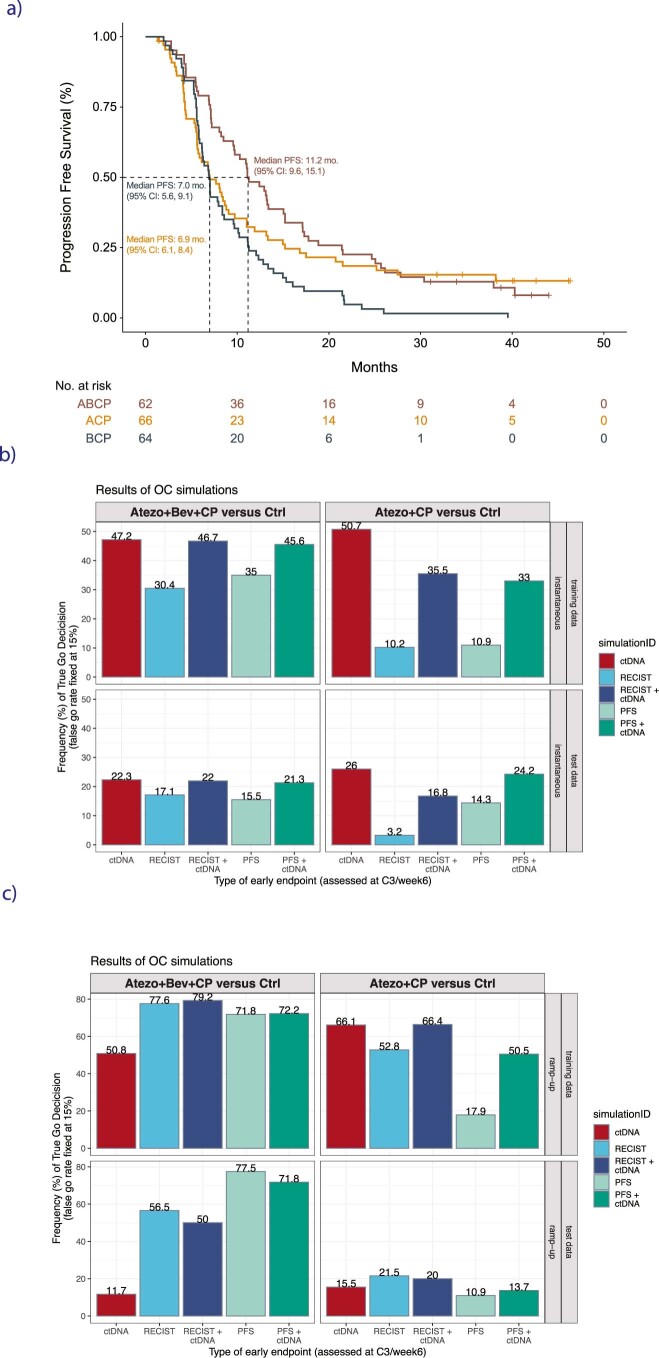
One of the great challenges in therapeutic oncology is determining who might achieve survival benefits from a particular therapy. Studies on longitudinal circulating tumor DNA (ctDNA) dynamics for the prediction of survival have generally been small or nonrandomized. We assessed ctDNA across 5 time points in 466 non-small-cell lung cancer (NSCLC) patients from the randomized phase 3 IMpower150 study comparing chemotherapy-immune checkpoint inhibitor (chemo-ICI) combinations and used machine learning to jointly model multiple ctDNA metrics to predict overall survival (OS). ctDNA assessments through cycle 3 day 1 of treatment enabled risk stratification of patients with stable disease (hazard ratio (HR) = 3.2 (2.0-5.3), P < 0.001; median 7.1 versus 22.3 months for high- versus low-intermediate risk) and with partial response (HR = 3.3 (1.7-6.4), P < 0.001; median 8.8 versus 28.6 months). The model also identified high-risk patients in an external validation cohort from the randomized phase 3 OAK study of ICI versus chemo in NSCLC (OS HR = 3.73 (1.83-7.60), P = 0.00012). Simulations of clinical trial scenarios employing our ctDNA model suggested that early ctDNA testing outperforms early radiographic imaging for predicting trial outcomes. Overall, measuring ctDNA dynamics during treatment can improve patient risk stratification and may allow early differentiation between competing therapies during clinical trials.
© 2023. The Author(s).
Conflict of interest statement
Z.J.A., D.S., K.S., C.C., Z.J.A., A.R., M.L., N.P., and W.Z. disclose current or recent employment with Roche. Z.J.A., D.S., K.S., G.O., C.C., M.L., D.F., N.P., W.Z., A.F., D.L., A.R., A.Y., and J.F. disclose stock or other ownership interests with Roche. G.O., A.Y., A.F., D.F., D.L., M.K., E.P., and J.F. disclose current or recent employment with FMI. M.R. discloses funding from Genentech, Pfizer, Spectrum, Takeda, Daiichi Sankyo, AstraZeneca, and Speaker Bureau for Genentech, AstraZeneca, Guardant, Jazz, Janssen and GI Therapeutics. M.S. discloses funding from Genentech, Pfizer, Spectrum, Takeda, Novartis, Beigene, AstraZeneca, and Daiichi Sankyo and discloses speaking fees from Genentech, Lilly, Blueprint, Guardant, BMS, Jazz, GI Therapeutics, Janssen and Amgen. M.L. discloses board member and shareholder in Foresight Diagnostics, scientific advisory board member and shareholder in Delfi Diagnostics and Prognomiq, Inc. The other authors declare no competing interests.
Figures
Comment in
-
High-resolution circulating tumor DNA testing predicts survival in metastatic lung cancer clinical trials.Nat Med. 2023 Apr;29(4):797-798. doi: 10.1038/s41591-023-02258-y. Nat Med. 2023. PMID: 37059837 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
