

Genetic association analysis of 77,539 genomes reveals rare disease etiologies
- PMID: 36928819
- PMCID: PMC10033407
- DOI: 10.1038/s41591-023-02211-z
Genetic association analysis of 77,539 genomes reveals rare disease etiologies
Abstract
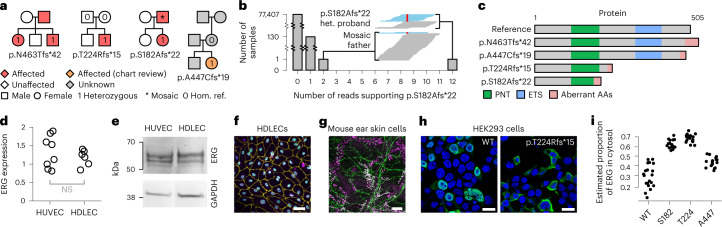
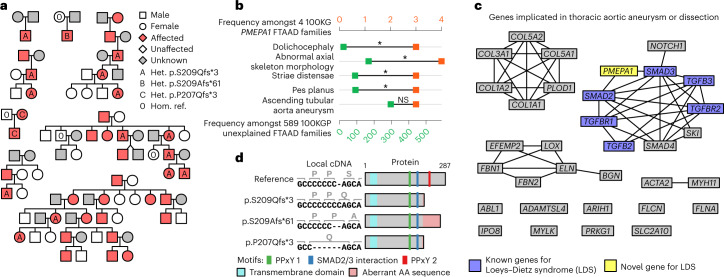
The genetic etiologies of more than half of rare diseases remain unknown. Standardized genome sequencing and phenotyping of large patient cohorts provide an opportunity for discovering the unknown etiologies, but this depends on efficient and powerful analytical methods. We built a compact database, the 'Rareservoir', containing the rare variant genotypes and phenotypes of 77,539 participants sequenced by the 100,000 Genomes Project. We then used the Bayesian genetic association method BeviMed to infer associations between genes and each of 269 rare disease classes assigned by clinicians to the participants. We identified 241 known and 19 previously unidentified associations. We validated associations with ERG, PMEPA1 and GPR156 by searching for pedigrees in other cohorts and using bioinformatic and experimental approaches. We provide evidence that (1) loss-of-function variants in the Erythroblast Transformation Specific (ETS)-family transcription factor encoding gene ERG lead to primary lymphoedema, (2) truncating variants in the last exon of transforming growth factor-β regulator PMEPA1 result in Loeys-Dietz syndrome and (3) loss-of-function variants in GPR156 give rise to recessive congenital hearing impairment. The Rareservoir provides a lightweight, flexible and portable system for synthesizing the genetic and phenotypic data required to study rare disease cohorts with tens of thousands of participants.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures











References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL161365/HL/NHLBI NIH HHS/United States
- RE/18/1/34212/BHF_/British Heart Foundation/United Kingdom
- MR/V037617/1/MRC_/Medical Research Council/United Kingdom
- 219506/Z/19/Z/WT_/Wellcome Trust/United Kingdom
- PG/17/33/32990/BHF_/British Heart Foundation/United Kingdom
- UL1 TR004419/TR/NCATS NIH HHS/United States
- PG/20/16/35047/BHF_/British Heart Foundation/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- MR/P011543/1/MRC_/Medical Research Council/United Kingdom
- R03 HD111492/HD/NICHD NIH HHS/United States
- R01 NS107428/NS/NINDS NIH HHS/United States
- R01 DC016295/DC/NIDCD NIH HHS/United States
- RG/17/7/33217/BHF_/British Heart Foundation/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
