

HDAC3 is critical in tumor development and therapeutic resistance in Kras-mutant non-small cell lung cancer
- PMID: 36930718
- PMCID: PMC10022903
- DOI: 10.1126/sciadv.add3243
HDAC3 is critical in tumor development and therapeutic resistance in Kras-mutant non-small cell lung cancer
Abstract
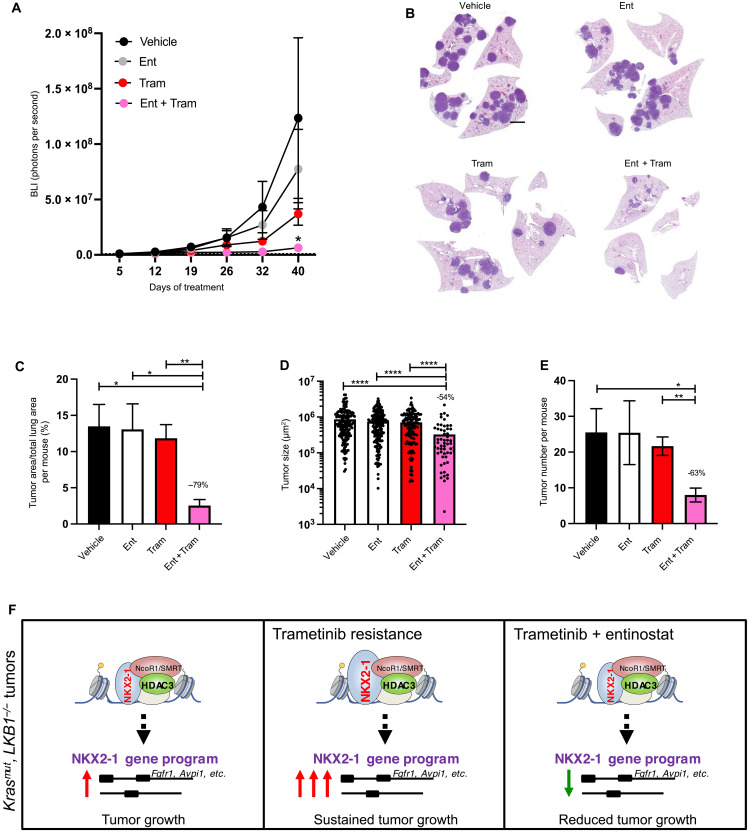
HDAC3 is one of the main targets of histone deacetylase (HDAC) inhibitors in clinical development as cancer therapies, yet the in vivo role of HDAC3 in solid tumors is unknown. We identified a critical role for HDAC3 in Kras-mutant lung cancer. Using genetically engineered mouse models (GEMMs), we found that HDAC3 is required for lung tumor growth in vivo. HDAC3 was found to direct and enhance the transcription effects of the lung cancer lineage transcription factor NKX2-1 to mediate expression of a common set of target genes. We identified FGFR1 as a critical previously unidentified target of HDAC3. Leveraging this, we identified that an HDAC3-dependent transcriptional cassette becomes hyperactivated as Kras/LKB1-mutant cells develop resistance to the MEK inhibitor trametinib, and this can be reversed by treatment with the HDAC1/HDAC3 inhibitor entinostat. We found that the combination of entinostat plus trametinib treatment elicits therapeutic benefit in the Kras/LKB1 GEMM.
Figures
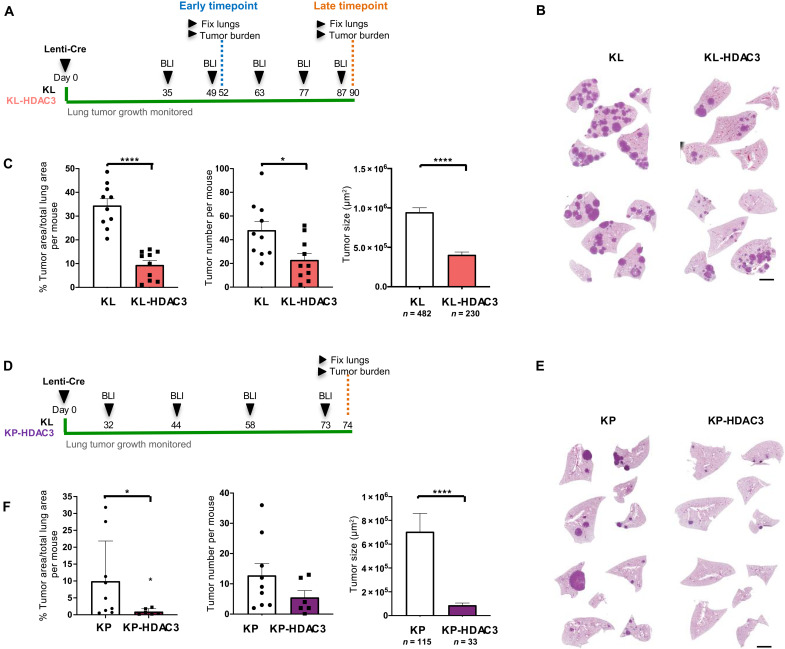
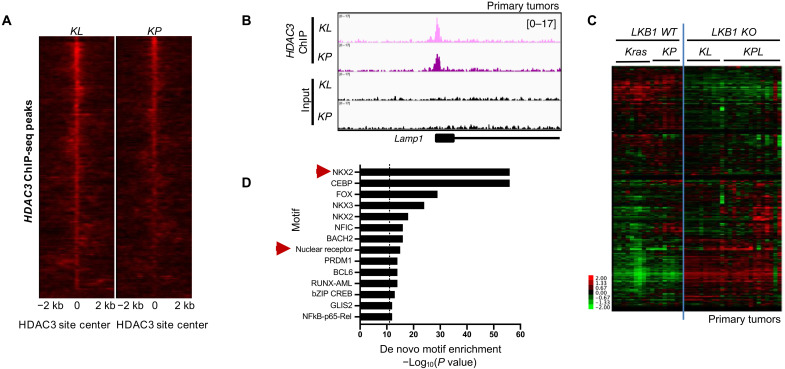
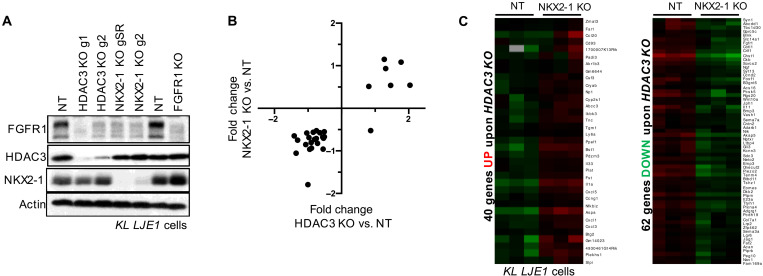
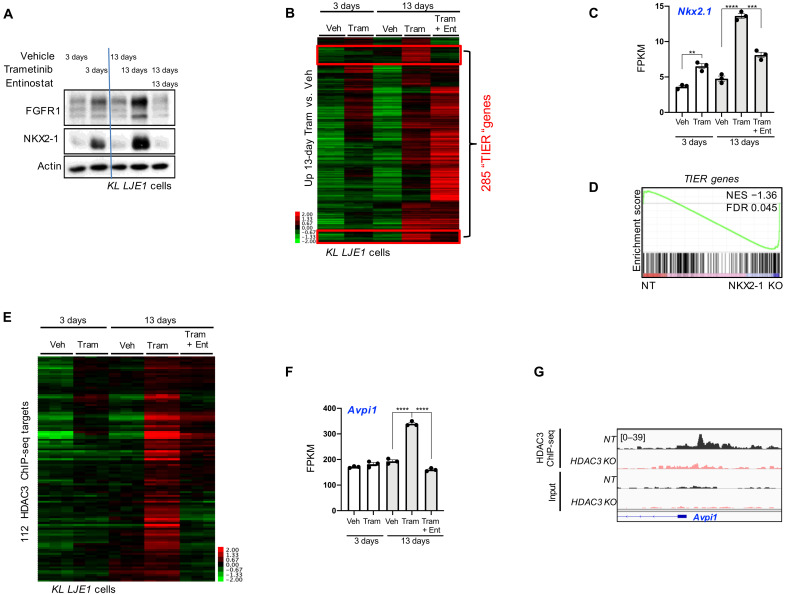
References
-
- T. De Raedt, E. Beert, E. Pasmant, A. Luscan, H. Brems, N. Ortonne, K. Helin, J. L. Hornick, V. Mautner, H. Kehrer-Sawatzki, W. Clapp, J. Bradner, M. Vidaud, M. Upadhyaya, E. Legius, K. Cichowski, PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514, 247–251 (2014). - PubMed
-
- N. Tasdemir, A. Banito, J. S. Roe, D. Alonso-Curbelo, M. Camiolo, D. F. Tschaharganeh, C. H. Huang, O. Aksoy, J. E. Bolden, C. C. Chen, M. Fennell, V. Thapar, A. Chicas, C. R. Vakoc, S. W. Lowe, BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 6, 612–629 (2016). - PMC - PubMed
-
- T. Shimamura, Z. Chen, M. Soucheray, J. Carretero, E. Kikuchi, J. H. Tchaicha, Y. Gao, K. A. Cheng, T. J. Cohoon, J. Qi, E. Akbay, A. C. Kimmelman, A. L. Kung, J. E. Bradner, K. K. Wong, Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin. Cancer Res. 19, 6183–6192 (2013). - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
