

Data integration across conditions improves turnover number estimates and metabolic predictions
- PMID: 36932067
- PMCID: PMC10023748
- DOI: 10.1038/s41467-023-37151-2
Data integration across conditions improves turnover number estimates and metabolic predictions
Abstract
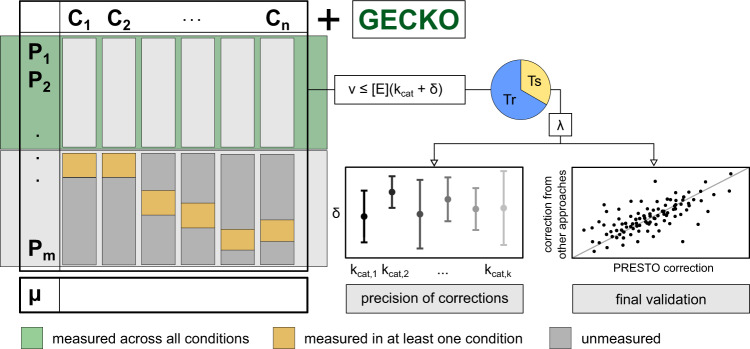
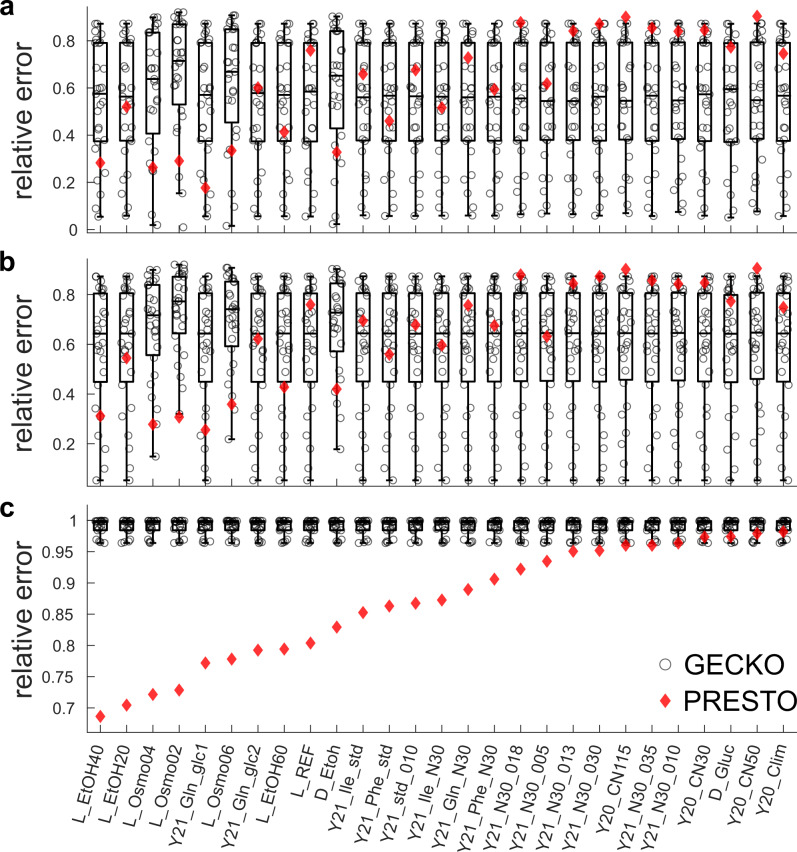
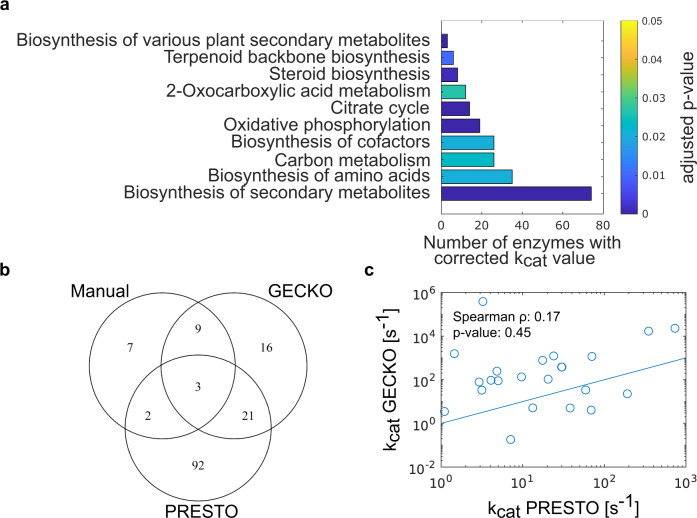
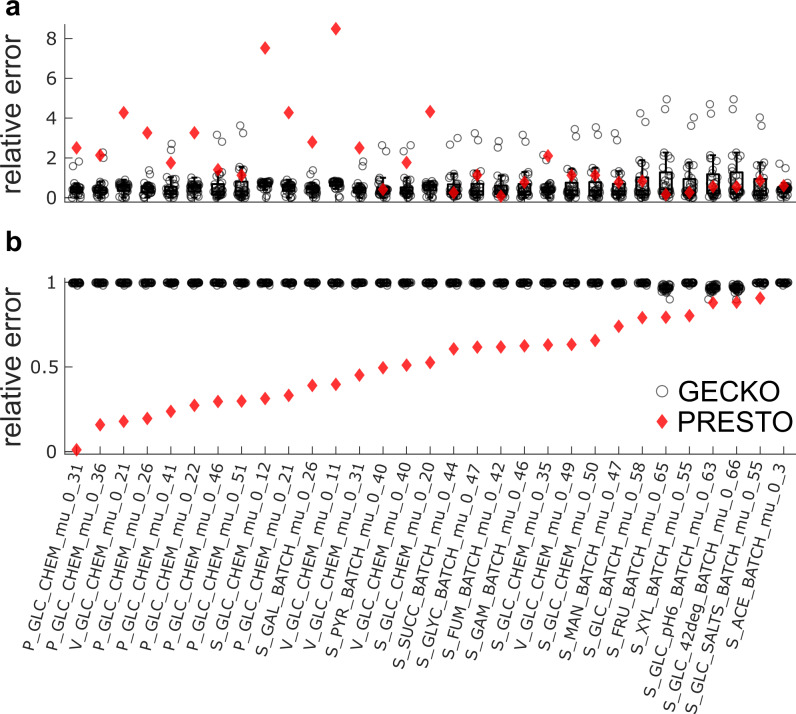
Turnover numbers characterize a key property of enzymes, and their usage in constraint-based metabolic modeling is expected to increase the prediction accuracy of diverse cellular phenotypes. In vivo turnover numbers can be obtained by integrating reaction rate and enzyme abundance measurements from individual experiments. Yet, their contribution to improving predictions of condition-specific cellular phenotypes remains elusive. Here, we show that available in vitro and in vivo turnover numbers lead to poor prediction of condition-specific growth rates with protein-constrained models of Escherichia coli and Saccharomyces cerevisiae, particularly when protein abundances are considered. We demonstrate that correction of turnover numbers by simultaneous consideration of proteomics and physiological data leads to improved predictions of condition-specific growth rates. Moreover, the obtained estimates are more precise than corresponding in vitro turnover numbers. Therefore, our approach provides the means to correct turnover numbers and paves the way towards cataloguing kcatomes of other organisms.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Chen Y, Nielsen J. Mathematical modeling of proteome constraints within metabolism. Curr. Opin. Syst. Biol. 2021;25:50–56. doi: 10.1016/j.coisb.2021.03.003. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
