

Diagnostic classification of childhood cancer using multiscale transcriptomics
- PMID: 36932241
- PMCID: PMC10033451
- DOI: 10.1038/s41591-023-02221-x
Diagnostic classification of childhood cancer using multiscale transcriptomics
Abstract
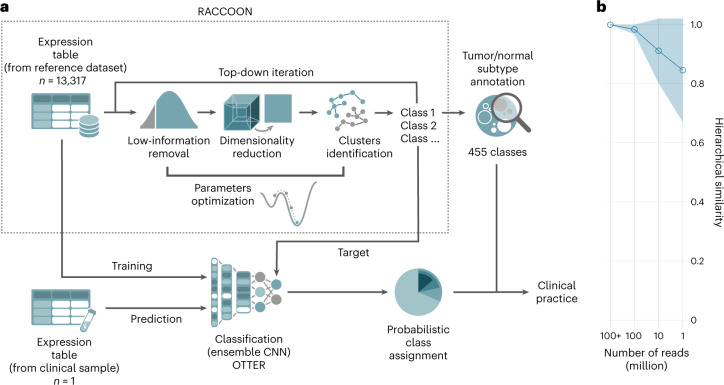
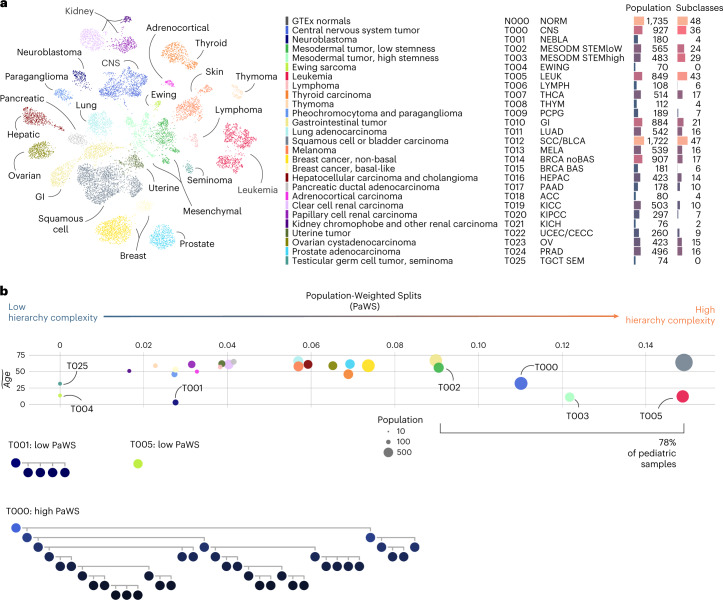
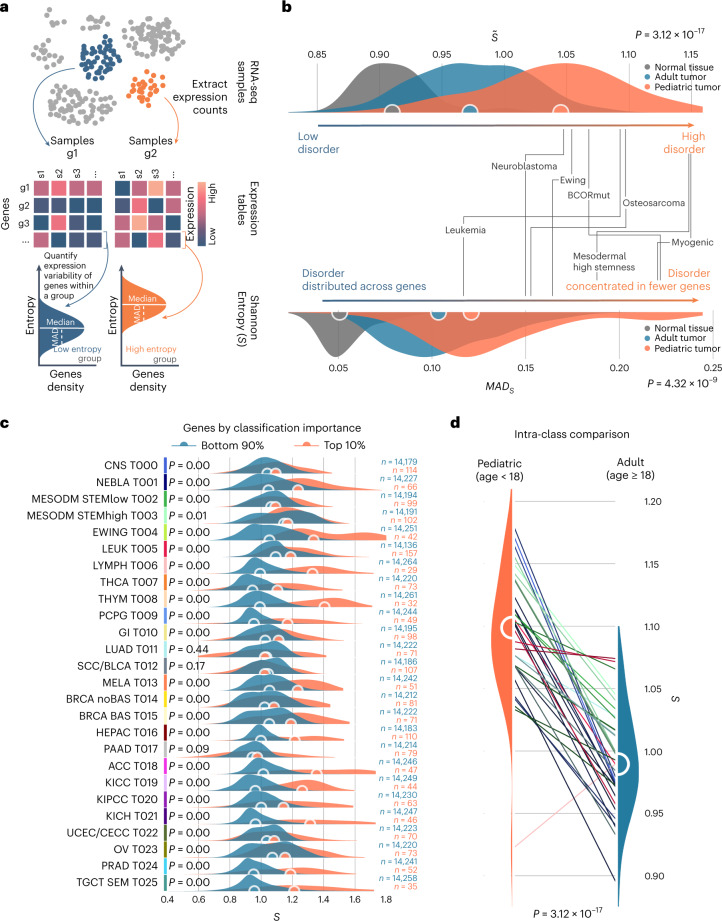
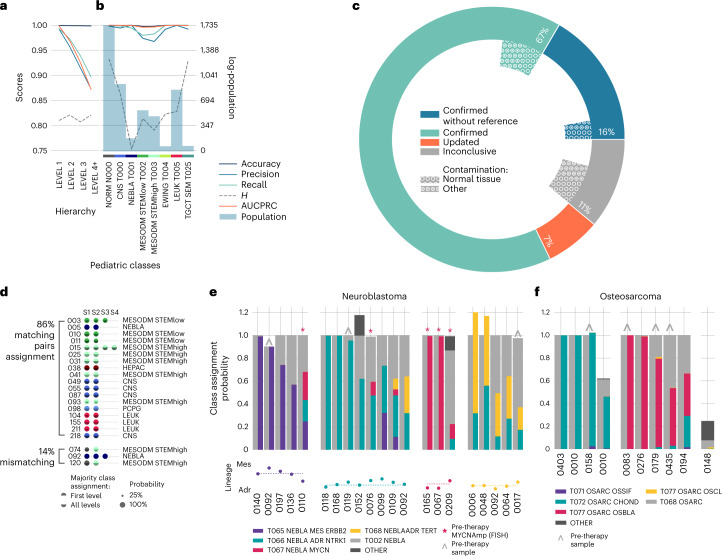
The causes of pediatric cancers' distinctiveness compared to adult-onset tumors of the same type are not completely clear and not fully explained by their genomes. In this study, we used an optimized multilevel RNA clustering approach to derive molecular definitions for most childhood cancers. Applying this method to 13,313 transcriptomes, we constructed a pediatric cancer atlas to explore age-associated changes. Tumor entities were sometimes unexpectedly grouped due to common lineages, drivers or stemness profiles. Some established entities were divided into subgroups that predicted outcome better than current diagnostic approaches. These definitions account for inter-tumoral and intra-tumoral heterogeneity and have the potential of enabling reproducible, quantifiable diagnostics. As a whole, childhood tumors had more transcriptional diversity than adult tumors, maintaining greater expression flexibility. To apply these insights, we designed an ensemble convolutional neural network classifier. We show that this tool was able to match or clarify the diagnosis for 85% of childhood tumors in a prospective cohort. If further validated, this framework could be extended to derive molecular definitions for all cancer types.
© 2023. The Author(s).
Conflict of interest statement
A.S. and F.C. report a filed patent application related to the use of transcriptional analysis to diagnose cancer and predict patient prognosis. The other authors declare no competing interests.
Figures











References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
