Large-scale analyses of CAV1 and CAV2 suggest their expression is higher in post-mortem ALS brain tissue and affects survival
- PMID: 36937187
- PMCID: PMC10017967
- DOI: 10.3389/fncel.2023.1112405
Large-scale analyses of CAV1 and CAV2 suggest their expression is higher in post-mortem ALS brain tissue and affects survival
Abstract
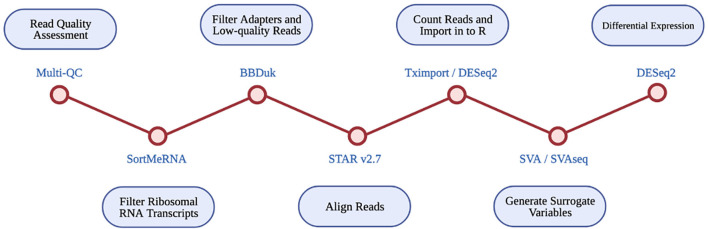
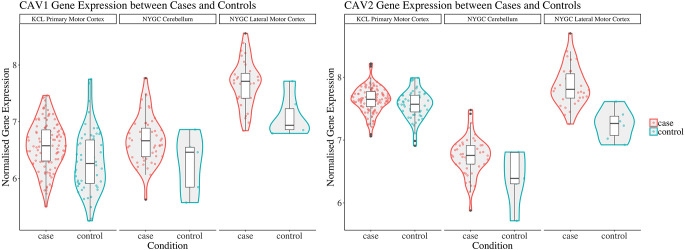
Introduction: Caveolin-1 and Caveolin-2 (CAV1 and CAV2) are proteins associated with intercellular neurotrophic signalling. There is converging evidence that CAV1 and CAV2 (CAV1/2) genes have a role in amyotrophic lateral sclerosis (ALS). Disease-associated variants have been identified within CAV1/2 enhancers, which reduce gene expression and lead to disruption of membrane lipid rafts. Methods: Using large ALS whole-genome sequencing and post-mortem RNA sequencing datasets (5,987 and 365 tissue samples, respectively), and iPSC-derived motor neurons from 55 individuals, we investigated the role of CAV1/2 expression and enhancer variants in the ALS phenotype. Results: We report a differential expression analysis between ALS cases and controls for CAV1 and CAV2 genes across various post-mortem brain tissues and three independent datasets. CAV1 and CAV2 expression was consistently higher in ALS patients compared to controls, with significant results across the primary motor cortex, lateral motor cortex, and cerebellum. We also identify increased survival among carriers of CAV1/2 enhancer mutations compared to non-carriers within Project MinE and slower progression as measured by the ALSFRS. Carriers showed a median increase in survival of 345 days. Discussion: These results add to an increasing body of evidence linking CAV1 and CAV2 genes to ALS. We propose that carriers of CAV1/2 enhancer mutations may be conceptualised as an ALS subtype who present a less severe ALS phenotype with a longer survival duration and slower progression. Upregulation of CAV1/2 genes in ALS cases may indicate a causal pathway or a compensatory mechanism. Given prior research supporting the beneficial role of CAV1/2 expression in ALS patients, we consider a compensatory mechanism to better fit the available evidence, although further investigation into the biological pathways associated with CAV1/2 is needed to support this conclusion.
Keywords: ALS (Amyotrophic lateral sclerosis); CAV1 and CAV2; Cav; caveolin; differential expression analysis (DEA); enhancer variant; neurodegeneration; survival analysis.
Copyright © 2023 Adey, Cooper-Knock, Al Khleifat, Fogh, van Damme, Corcia, Couratier, Hardiman, McLaughlin, Gotkine, Drory, Silani, Ticozzi, Veldink, van den Berg, de Carvalho, Pinto, Mora Pardina, Povedano Panades, Andersen, Weber, Başak, Shaw, Shaw, Morrison, Landers, Glass, Vourc’h, Dobson, Breen, Al-Chalabi, Jones and Iacoangeli.
Conflict of interest statement
JV reports to have sponsored research agreements with Biogen and Astra Zeneca. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures