

A new method to synthesize multiple gRNA libraries and functional mapping of mammalian H3K4me3 regions
- PMID: 36938898
- PMCID: PMC10201432
- DOI: 10.1093/nar/gkad198
A new method to synthesize multiple gRNA libraries and functional mapping of mammalian H3K4me3 regions
Abstract
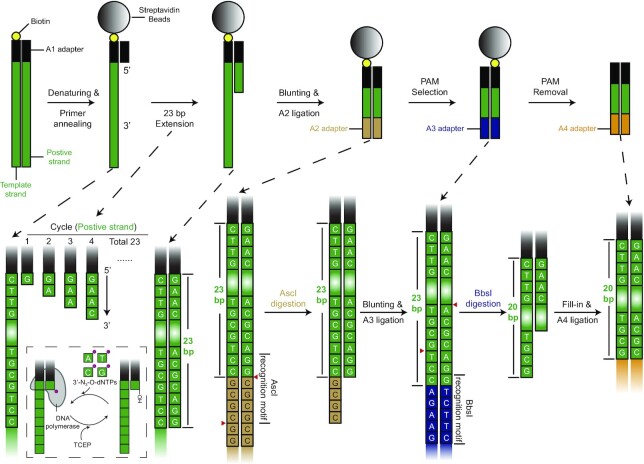
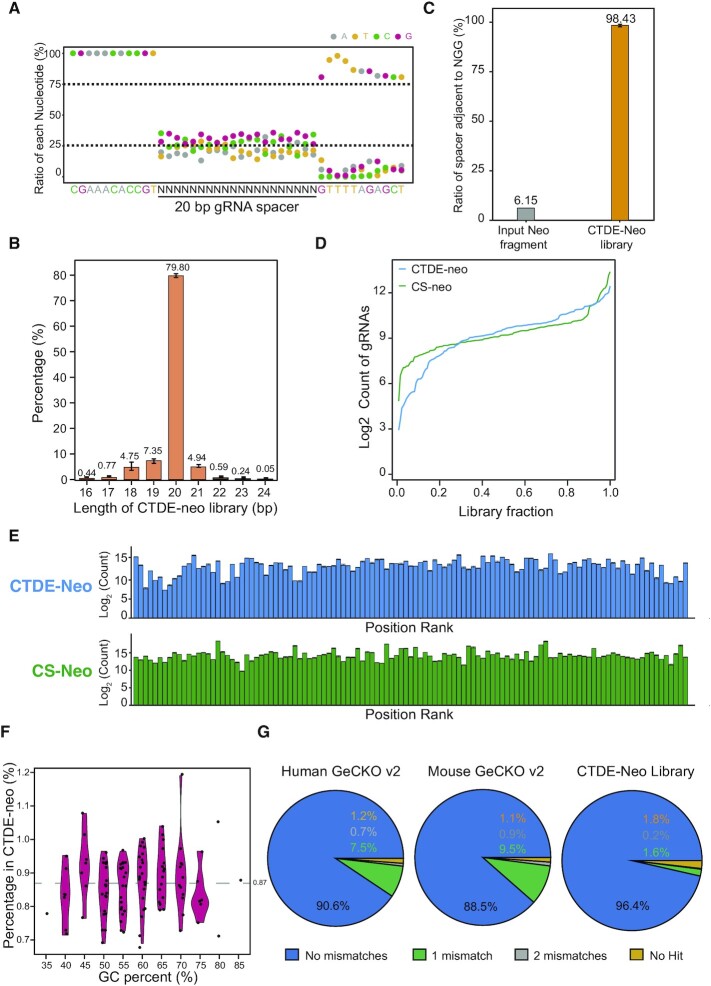
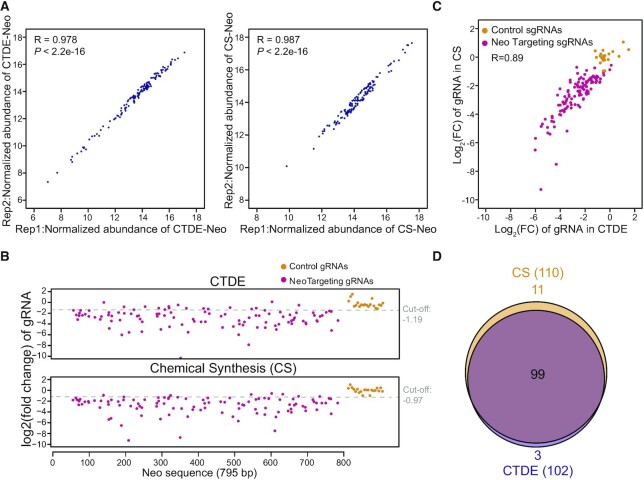
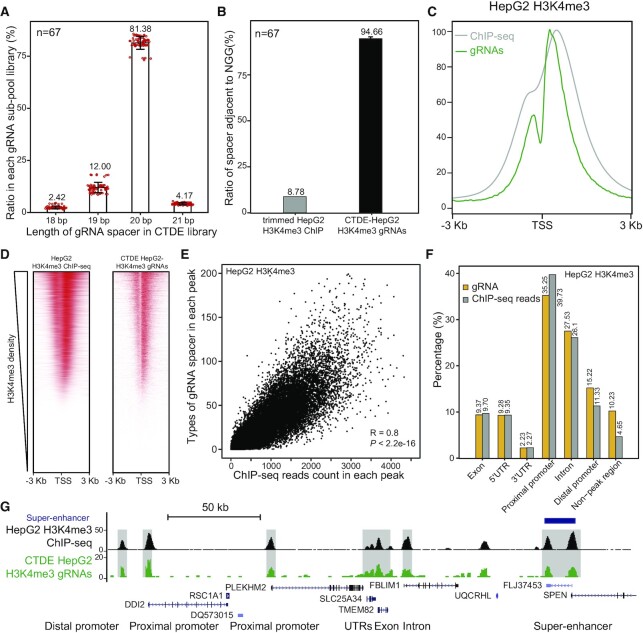
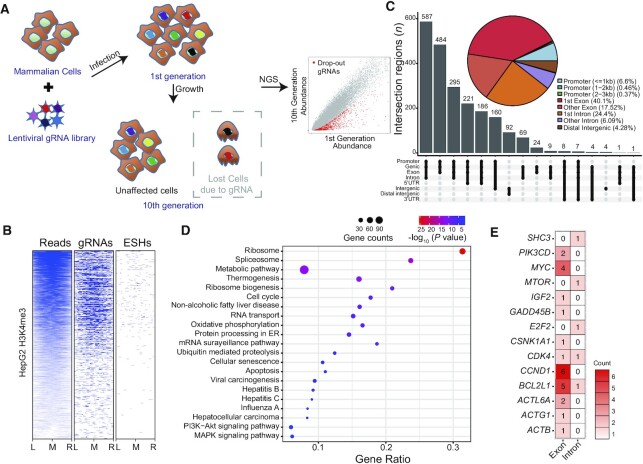
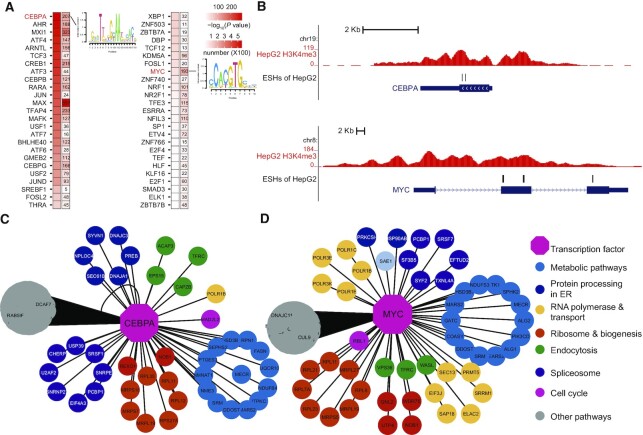
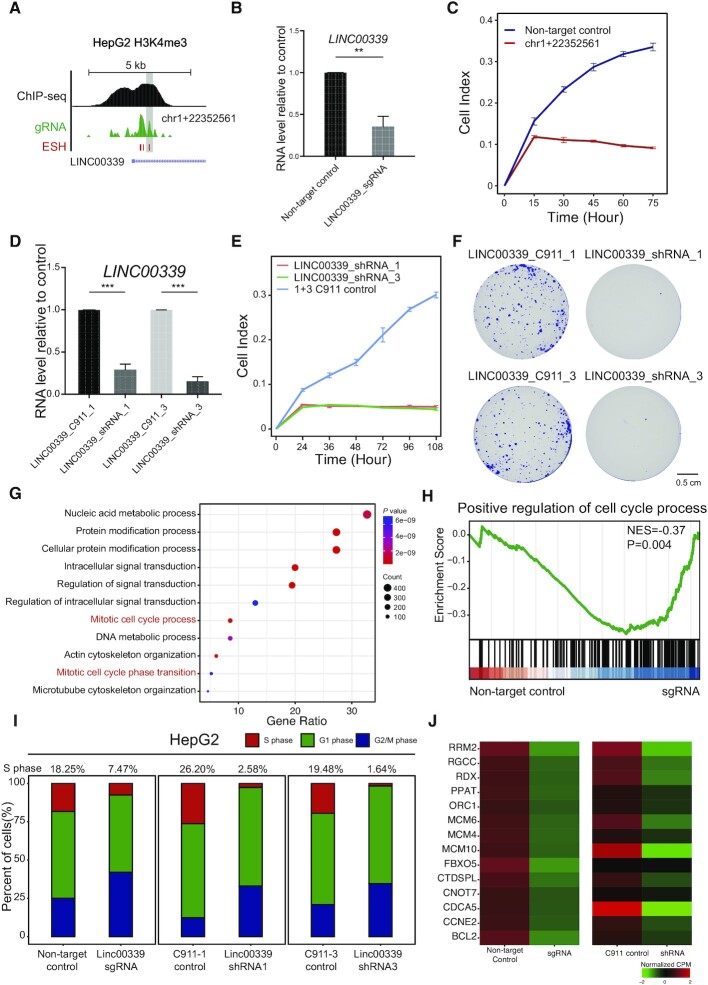
Genetic screening based on the clustered regularly interspaced palindromic repeat (CRISPR) system has been indicated to be a powerful tool for identifying regulatory genes or cis-elements. However, when applying CRISPR screens to pinpoint functional elements at particular loci, a large number of guide RNA (gRNA) spacers may be required to achieve saturated coverage. Here, we present a controlled template-dependent elongation (CTDE) method relying on reversible terminators to synthesize gRNA libraries with genomic regions of interest. By applying this approach to H3K4me3 chromatin immunoprecipitation (ChIP)-derived DNA of mammalian cells, mega-sized gRNA libraries were synthesized in a tissue-specific manner, with which we conducted screening experiments to annotate essential sites for cell proliferation. Additionally, we confirmed that an essential site within the intron of LINC00339 regulates its own mRNA and that LINC00339 is a novel regulator of the cell cycle that maintains HepG2 proliferation. The CTDE method has the potential to be automated with high efficiency at low cost, and will be widely used to identify functional elements in mammalian genomes.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
