

prolfqua: A Comprehensive R-Package for Proteomics Differential Expression Analysis
- PMID: 36939687
- PMCID: PMC10088014
- DOI: 10.1021/acs.jproteome.2c00441
prolfqua: A Comprehensive R-Package for Proteomics Differential Expression Analysis
Abstract
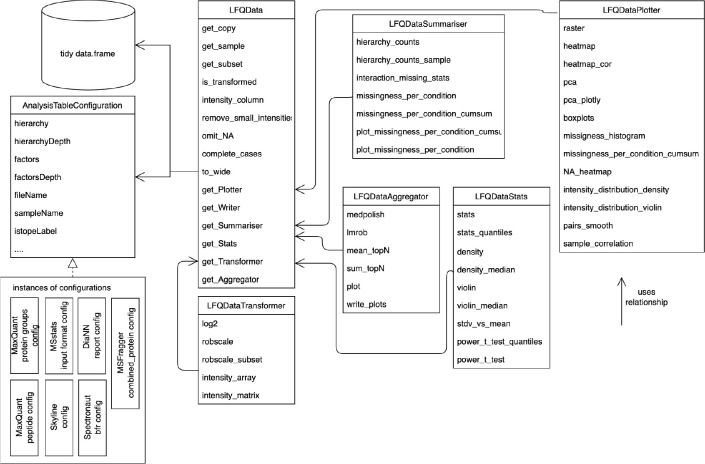
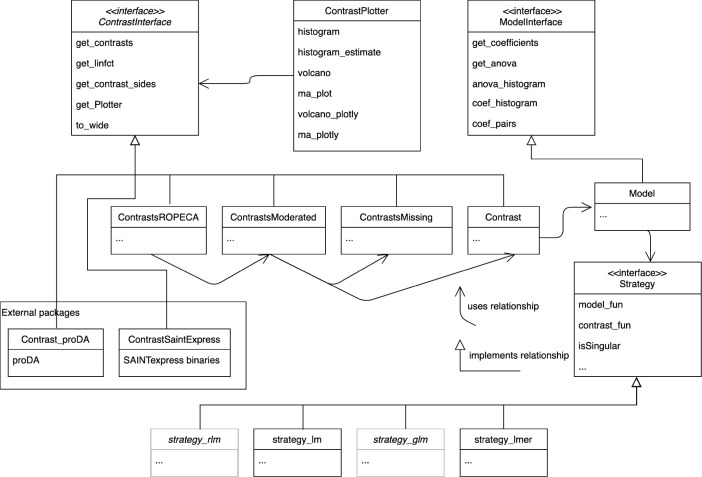
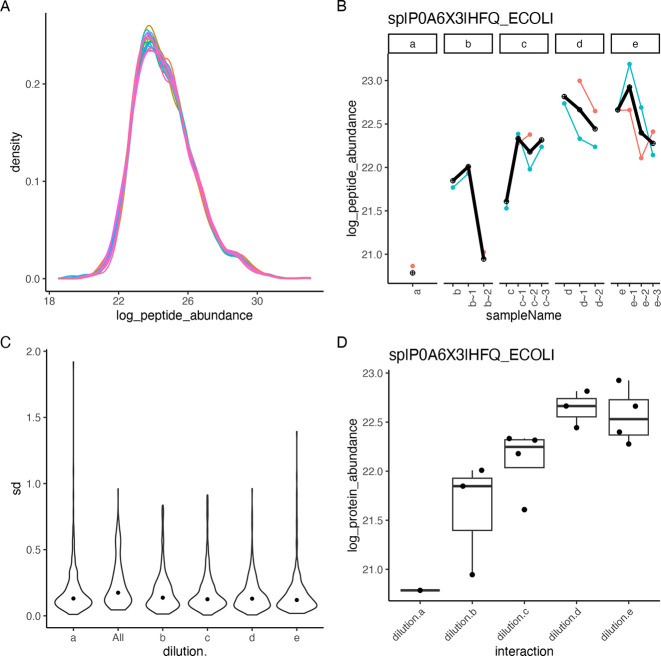
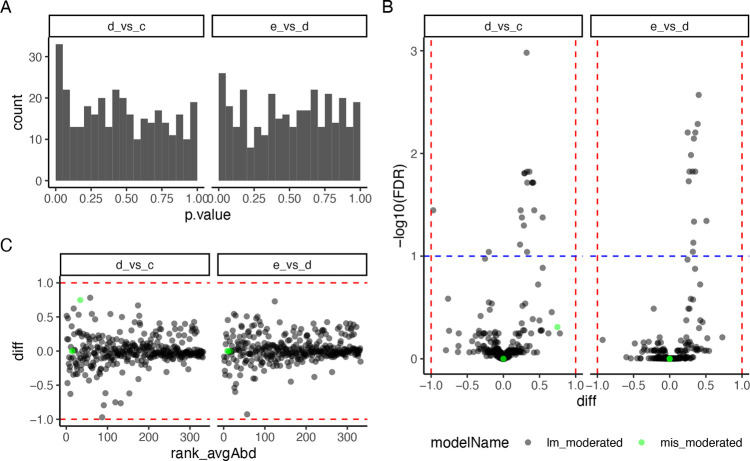
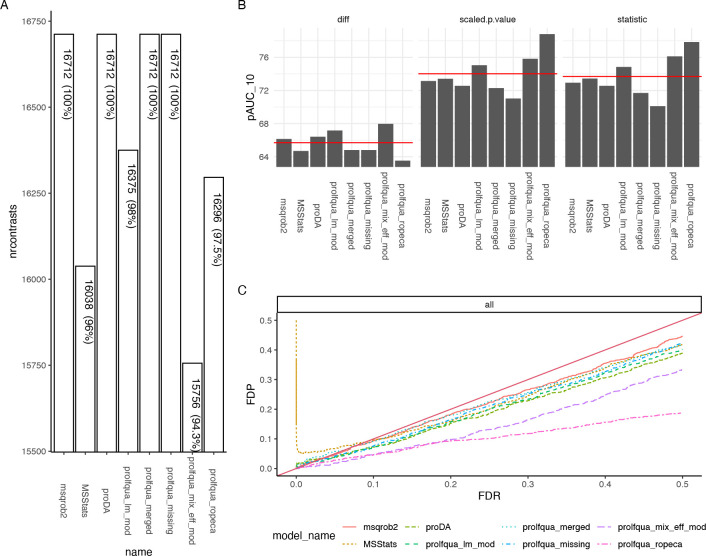
Mass spectrometry is widely used for quantitative proteomics studies, relative protein quantification, and differential expression analysis of proteins. There is a large variety of quantification software and analysis tools. Nevertheless, there is a need for a modular, easy-to-use application programming interface in R that transparently supports a variety of well principled statistical procedures to make applying them to proteomics data, comparing and understanding their differences easy. The prolfqua package integrates essential steps of the mass spectrometry-based differential expression analysis workflow: quality control, data normalization, protein aggregation, statistical modeling, hypothesis testing, and sample size estimation. The package makes integrating new data formats easy. It can be used to model simple experimental designs with a single explanatory variable and complex experiments with multiple factors and hypothesis testing. The implemented methods allow sensitive and specific differential expression analysis. Furthermore, the package implements benchmark functionality that can help to compare data acquisition, data preprocessing, or data modeling methods using a gold standard data set. The application programmer interface of prolfqua strives to be clear, predictable, discoverable, and consistent to make proteomics data analysis application development easy and exciting. Finally, the prolfqua R-package is available on GitHub https://github.com/fgcz/prolfqua, distributed under the MIT license. It runs on all platforms supported by the R free software environment for statistical computing and graphics.
Keywords: differential expression analysis; proteomics; statistical software.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
