

Dissection of complement and Fc-receptor-mediated pathomechanisms of autoantibodies to myelin oligodendrocyte glycoprotein
- PMID: 36943883
- PMCID: PMC10068779
- DOI: 10.1073/pnas.2300648120
Dissection of complement and Fc-receptor-mediated pathomechanisms of autoantibodies to myelin oligodendrocyte glycoprotein
Abstract
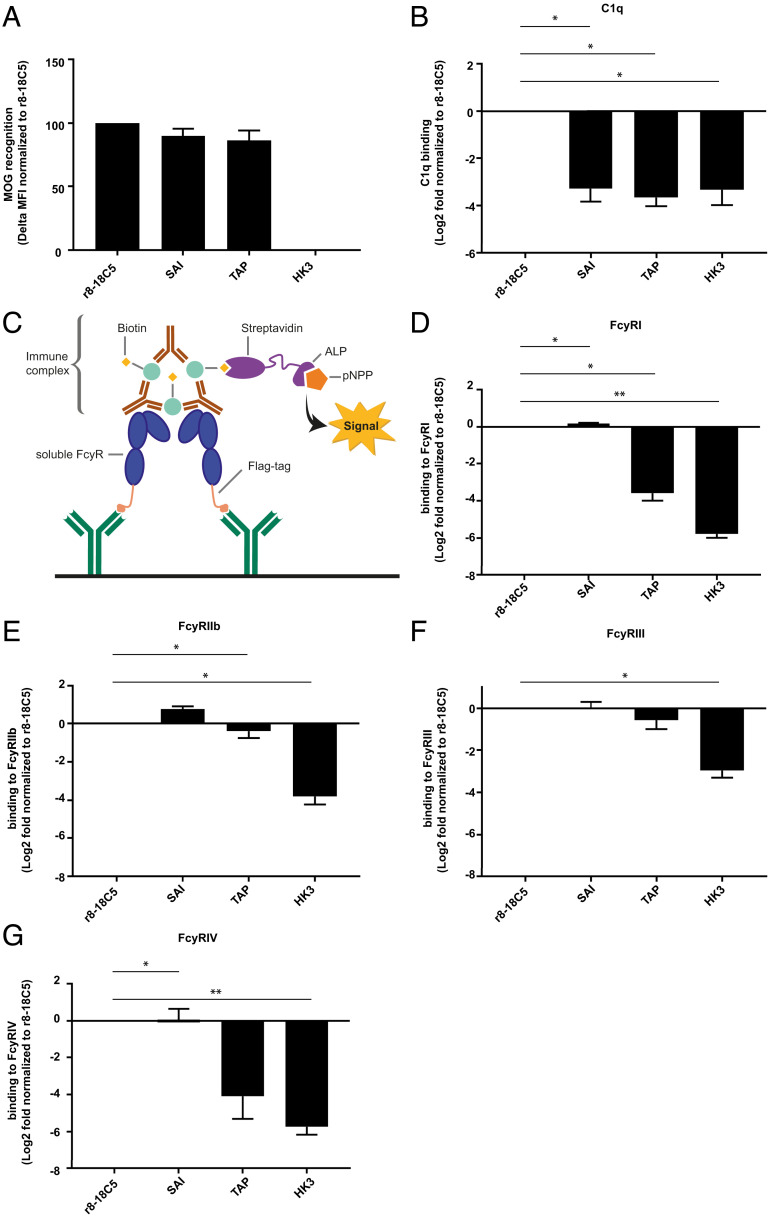
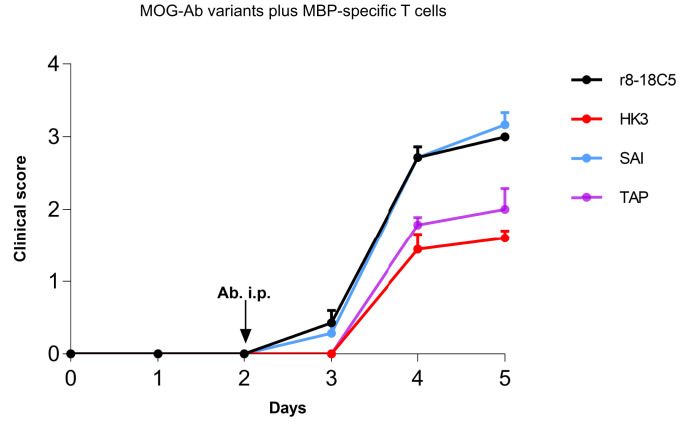
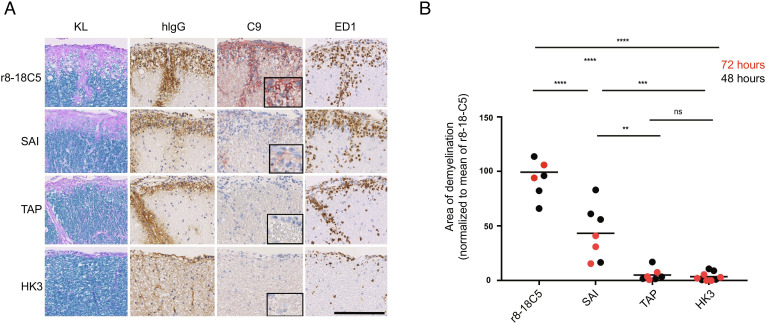
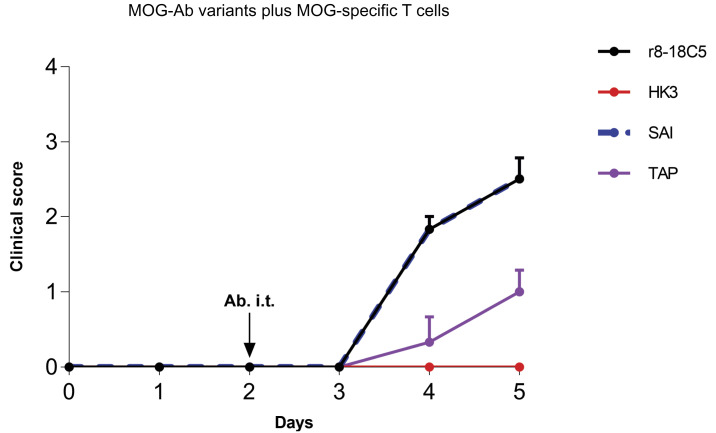
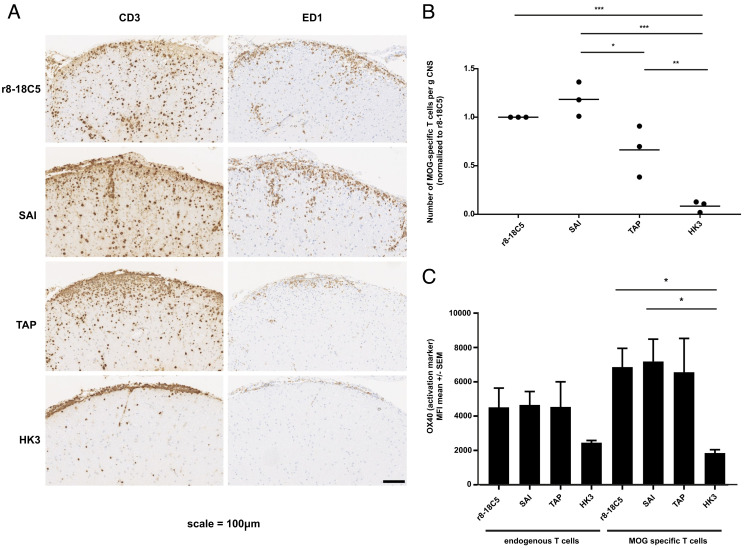
Autoantibodies against myelin oligodendrocyte glycoprotein (MOG) have recently been established to define a new disease entity, MOG-antibody-associated disease (MOGAD), which is clinically overlapping with multiple sclerosis. MOG-specific antibodies (Abs) from patients are pathogenic, but the precise effector mechanisms are currently still unknown and no therapy is approved for MOGAD. Here, we determined the contributions of complement and Fc-receptor (FcR)-mediated effects in the pathogenicity of MOG-Abs. Starting from a recombinant anti-MOG (mAb) with human IgG1 Fc, we established MOG-specific mutant mAbs with differential FcR and C1q binding. We then applied selected mutants of this MOG-mAb in two animal models of experimental autoimmune encephalomyelitis. First, we found MOG-mAb-induced demyelination was mediated by both complement and FcRs about equally. Second, we found that MOG-Abs enhanced activation of cognate MOG-specific T cells in the central nervous system (CNS), which was dependent on FcR-, but not C1q-binding. The identification of complement-dependent and -independent pathomechanisms of MOG-Abs has implications for therapeutic strategies in MOGAD.
Keywords: Autoimmunity; demyelination; effector mechanisms; inflammation; neuroimmunology.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
