

Benchmarking integration of single-cell differential expression
- PMID: 36944632
- PMCID: PMC10030080
- DOI: 10.1038/s41467-023-37126-3
Benchmarking integration of single-cell differential expression
Abstract
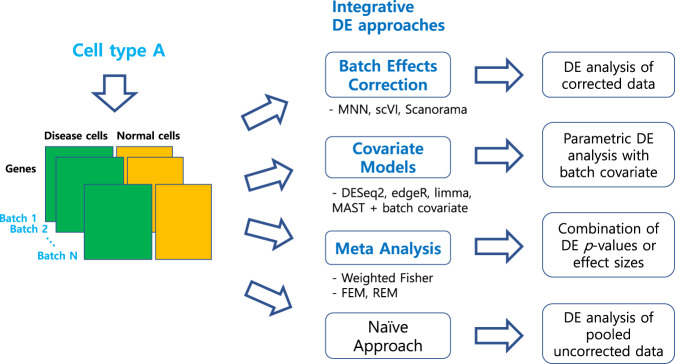
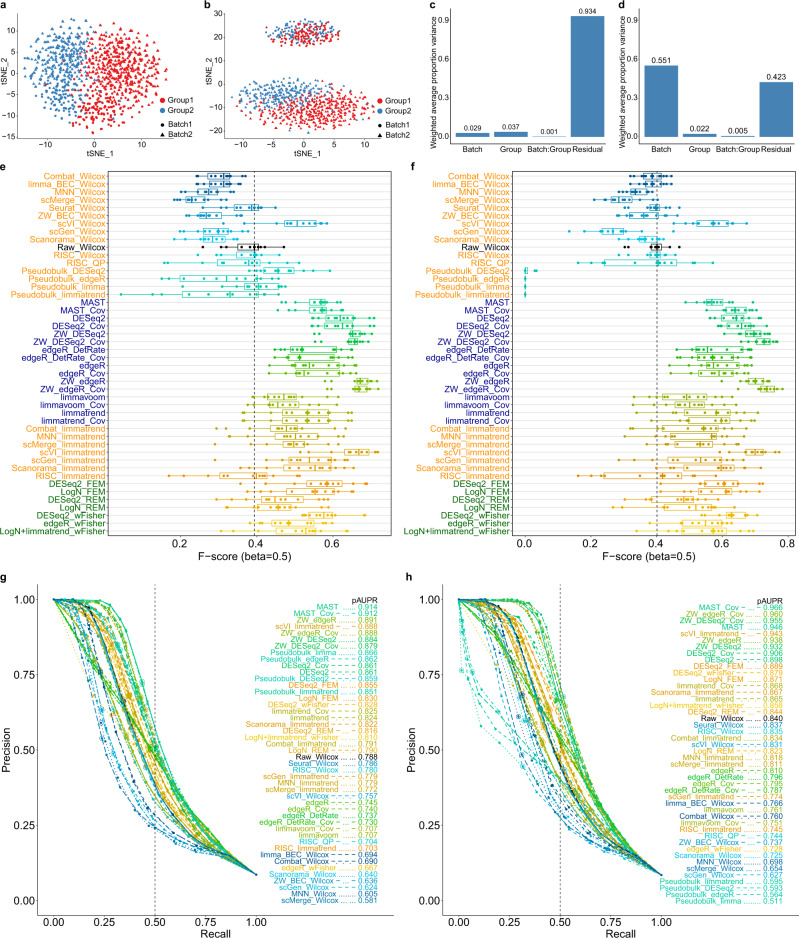
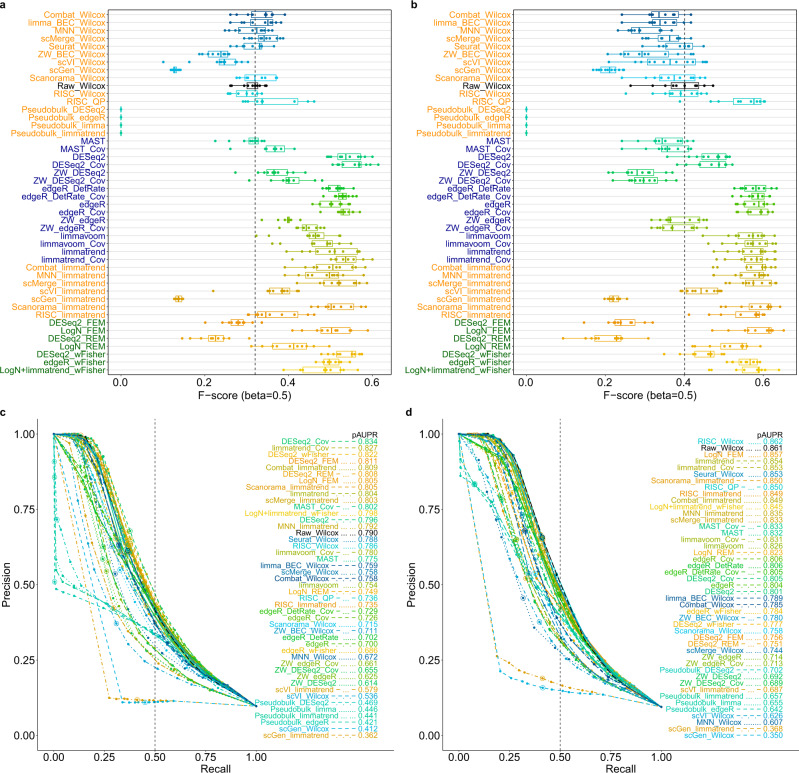
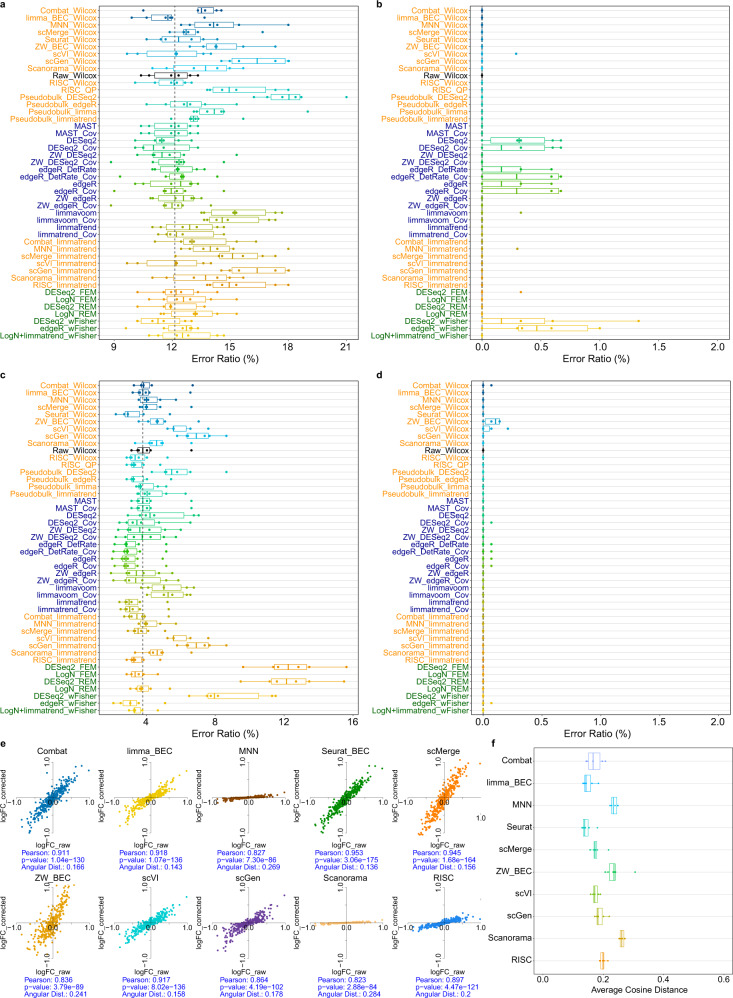
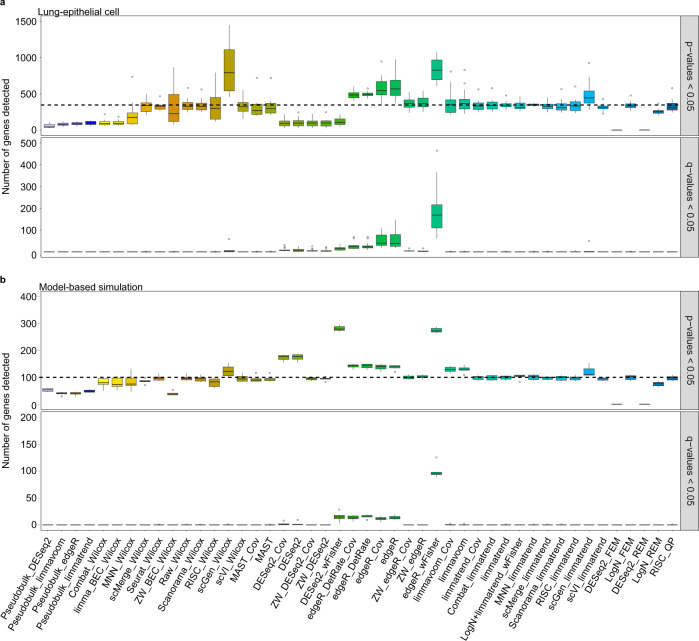
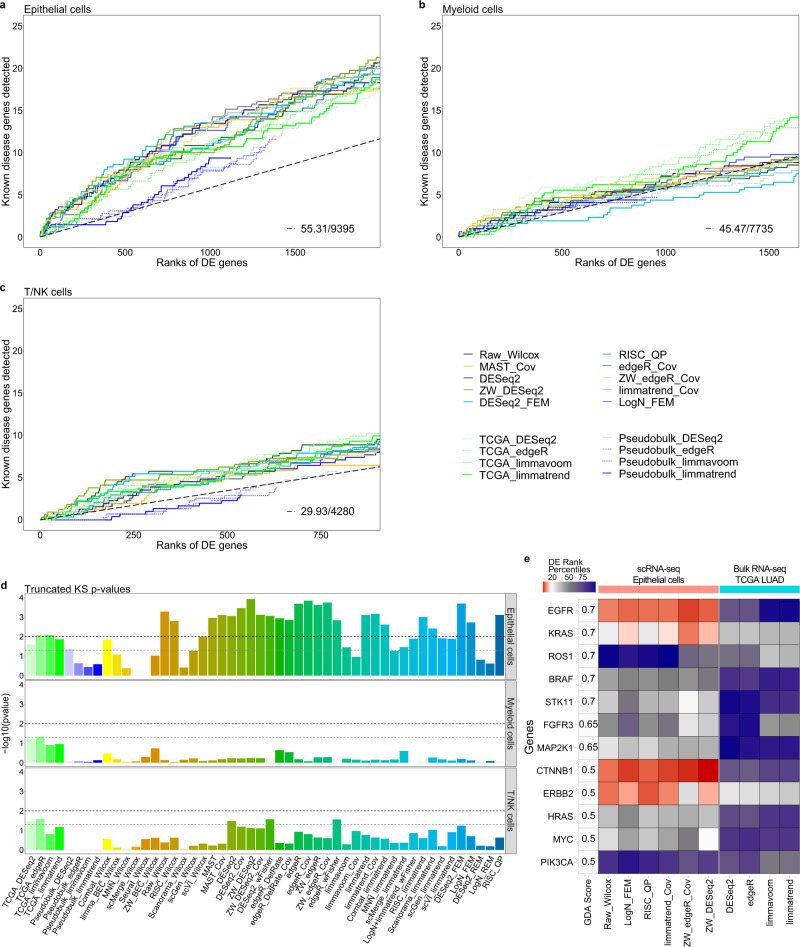
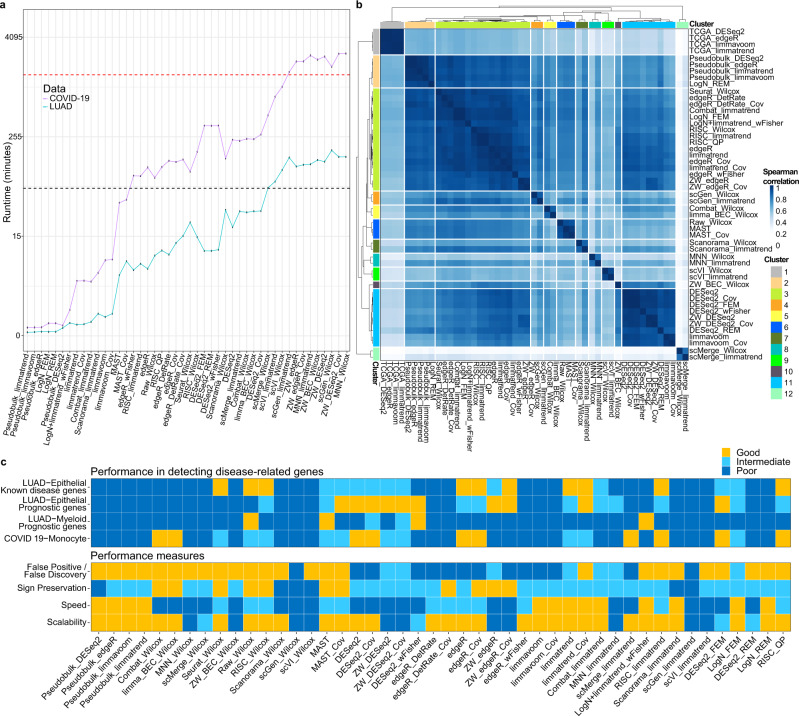
Integration of single-cell RNA sequencing data between different samples has been a major challenge for analyzing cell populations. However, strategies to integrate differential expression analysis of single-cell data remain underinvestigated. Here, we benchmark 46 workflows for differential expression analysis of single-cell data with multiple batches. We show that batch effects, sequencing depth and data sparsity substantially impact their performances. Notably, we find that the use of batch-corrected data rarely improves the analysis for sparse data, whereas batch covariate modeling improves the analysis for substantial batch effects. We show that for low depth data, single-cell techniques based on zero-inflation model deteriorate the performance, whereas the analysis of uncorrected data using limmatrend, Wilcoxon test and fixed effects model performs well. We suggest several high-performance methods under different conditions based on various simulation and real data analyses. Additionally, we demonstrate that differential expression analysis for a specific cell type outperforms that of large-scale bulk sample data in prioritizing disease-related genes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
