

This is a preprint.
Variants in ACTC1 underlie distal arthrogryposis accompanied by congenital heart defects
- PMID: 36945405
- PMCID: PMC10029015
- DOI: 10.1101/2023.03.07.23286862
Variants in ACTC1 underlie distal arthrogryposis accompanied by congenital heart defects
Update in
-
Variants in ACTC1 underlie distal arthrogryposis accompanied by congenital heart defects.HGG Adv. 2023 Jun 15;4(3):100213. doi: 10.1016/j.xhgg.2023.100213. eCollection 2023 Jul 13. HGG Adv. 2023. PMID: 37457373 Free PMC article.
Abstract
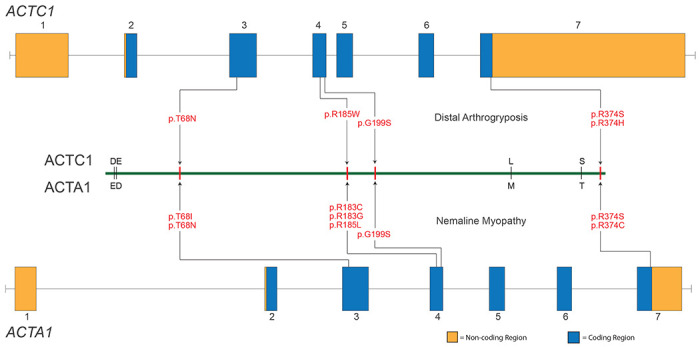
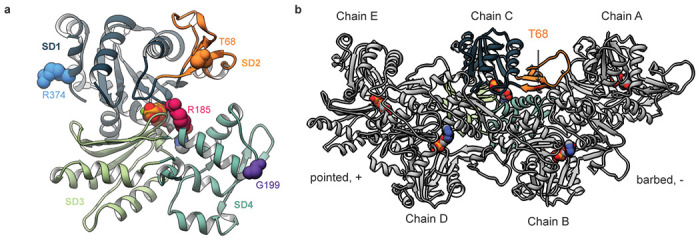
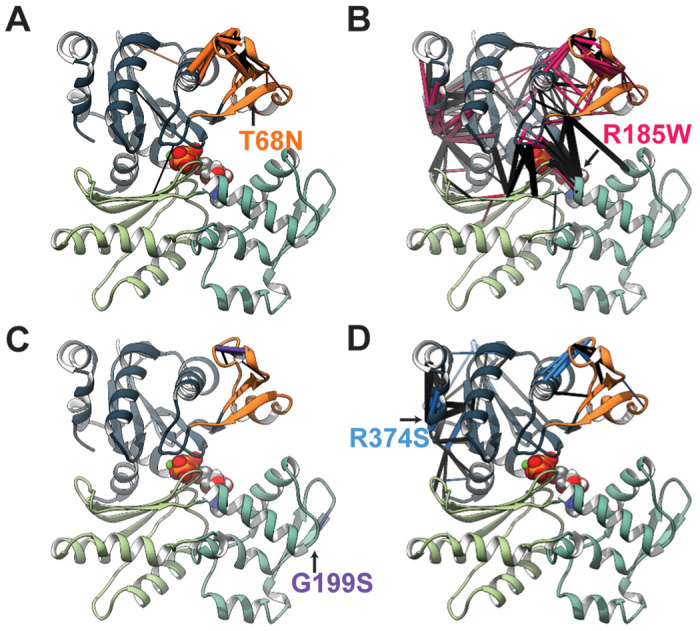
Contraction of the human sarcomere is the result of interactions between myosin cross-bridges and actin filaments. Pathogenic variants in genes such as MYH7 , TPM1 , and TNNI3 that encode parts of the cardiac sarcomere cause muscle diseases that affect the heart, such as dilated cardiomyopathy and hypertrophic cardiomyopathy. In contrast, pathogenic variants in homologous genes MYH2 , TPM2 , and TNNI2 , that encode parts of the skeletal muscle sarcomere, cause muscle diseases affecting skeletal muscle, such as the distal arthrogryposis (DA) syndromes and skeletal myopathies. To date, there have been few reports of genes (e.g., MYH7 ) encoding sarcomeric proteins in which the same pathogenic variant affects both skeletal and cardiac muscle. Moreover, none of the known genes underlying DA have been found to contain mutations that also cause cardiac abnormalities. We report five families with DA due to heterozygous missense variants in the gene actin, alpha, cardiac muscle 1 ( ACTC1 ). ACTC1 encodes a highly conserved actin that binds to myosin in both cardiac and skeletal muscle. Mutations in ACTC1 have previously been found to underlie atrial septal defect, dilated cardiomyopathy, hypertrophic cardiomyopathy, and left ventricular noncompaction. Our discovery delineates a new DA condition due to mutations in ACTC1 and suggests that some functions of actin, alpha, cardiac muscle 1 are shared in cardiac and skeletal muscle.
Conflict of interest statement
Declaration of Interests
MJB and JXC are the Editor-in-Chief and Deputy Editor of
Figures


References
-
- Lindskog C., Linné J., Fagerberg L., Hallström B.M., Sundberg C.J., Lindholm M., Huss M., Kampf C., Choi H., Liem D.A., et al. (2015). The human cardiac and skeletal muscle proteomes defined by transcriptomics and antibody-based profiling. Bmc Genomics 16, 475. 10.1186/s12864-015-1686-y. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous