

This is a preprint.
A deep population reference panel of tandem repeat variation
- PMID: 36945429
- PMCID: PMC10028971
- DOI: 10.1101/2023.03.09.531600
A deep population reference panel of tandem repeat variation
Update in
-
A deep population reference panel of tandem repeat variation.Nat Commun. 2023 Oct 23;14(1):6711. doi: 10.1038/s41467-023-42278-3. Nat Commun. 2023. PMID: 37872149 Free PMC article.
Abstract
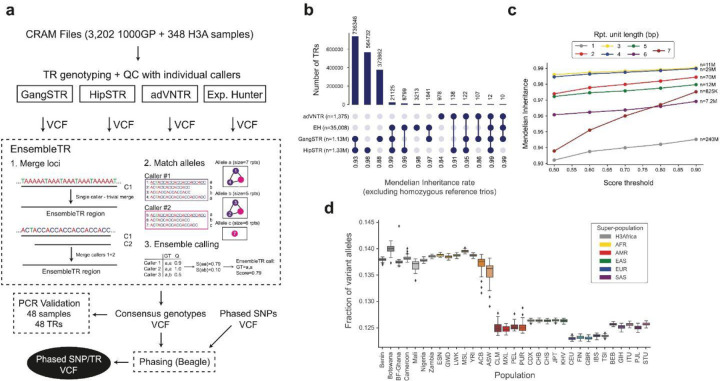
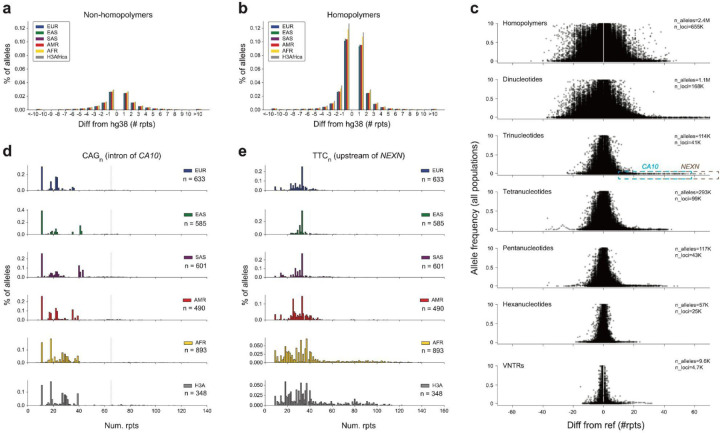
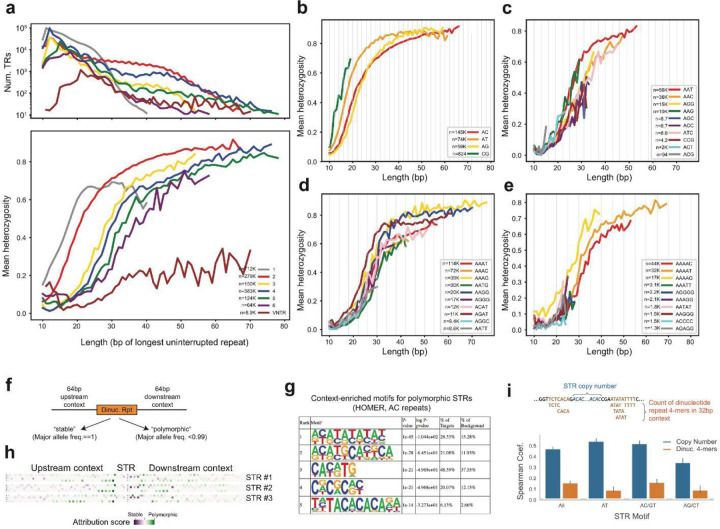
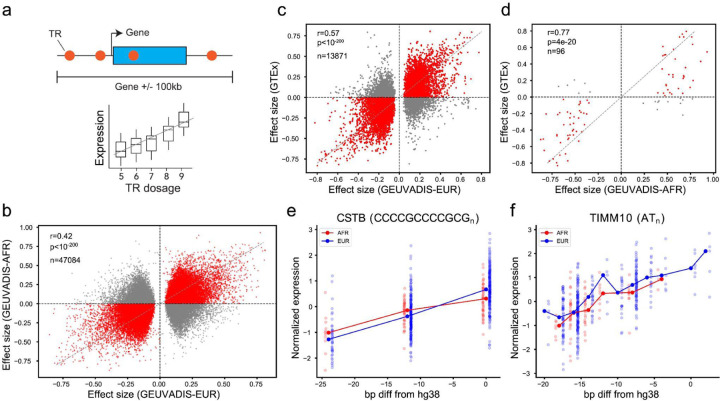
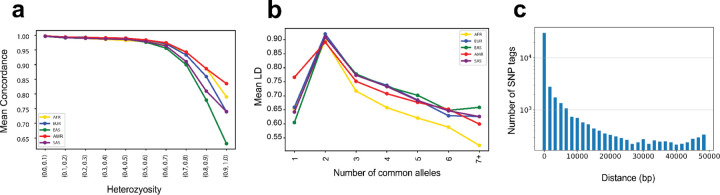
Tandem repeats (TRs) represent one of the largest sources of genetic variation in humans and are implicated in a range of phenotypes. Here we present a deep characterization of TR variation based on high coverage whole genome sequencing from 3,550 diverse individuals from the 1000 Genomes Project and H3Africa cohorts. We develop a method, EnsembleTR, to integrate genotypes from four separate methods resulting in high-quality genotypes at more than 1.7 million TR loci. Our catalog reveals novel sequence features influencing TR heterozygosity, identifies population-specific trinucleotide expansions, and finds hundreds of novel eQTL signals. Finally, we generate a phased haplotype panel which can be used to impute most TRs from nearby single nucleotide polymorphisms (SNPs) with high accuracy. Overall, the TR genotypes and reference haplotype panel generated here will serve as valuable resources for future genome-wide and population-wide studies of TRs and their role in human phenotypes.
Conflict of interest statement
Competing interests V.B. is a co-founder, consultant, SAB member and has equity interest in Boundless Bio, inc. and Abterra, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Figures
References
-
- Weber J. L. & Wong C. Mutation of human short tandem repeats. Hum. Mol. Genet. 2, 1123–1128 (1993). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources