

This is a preprint.
Structurally divergent and recurrently mutated regions of primate genomes
- PMID: 36945442
- PMCID: PMC10028934
- DOI: 10.1101/2023.03.07.531415
Structurally divergent and recurrently mutated regions of primate genomes
Update in
-
Structurally divergent and recurrently mutated regions of primate genomes.Cell. 2024 Mar 14;187(6):1547-1562.e13. doi: 10.1016/j.cell.2024.01.052. Epub 2024 Feb 29. Cell. 2024. PMID: 38428424 Free PMC article.
Abstract
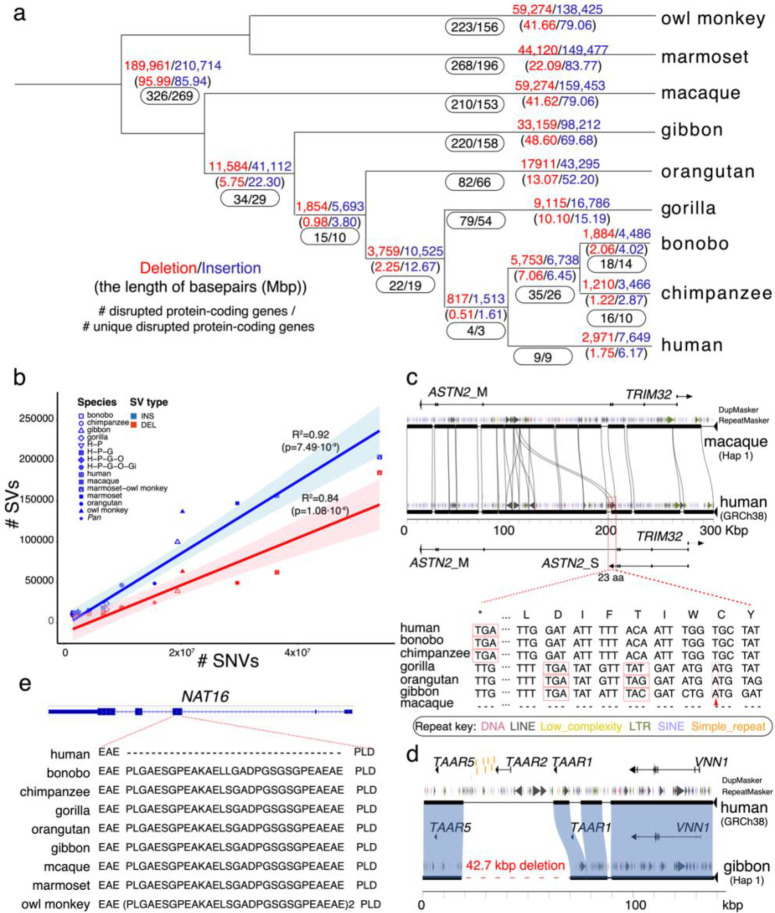
To better understand the pattern of primate genome structural variation, we sequenced and assembled using multiple long-read sequencing technologies the genomes of eight nonhuman primate species, including New World monkeys (owl monkey and marmoset), Old World monkey (macaque), Asian apes (orangutan and gibbon), and African ape lineages (gorilla, bonobo, and chimpanzee). Compared to the human genome, we identified 1,338,997 lineage-specific fixed structural variants (SVs) disrupting 1,561 protein-coding genes and 136,932 regulatory elements, including the most complete set of human-specific fixed differences. Across 50 million years of primate evolution, we estimate that 819.47 Mbp or ~27% of the genome has been affected by SVs based on analysis of these primate lineages. We identify 1,607 structurally divergent regions (SDRs) wherein recurrent structural variation contributes to creating SV hotspots where genes are recurrently lost (CARDs, ABCD7, OLAH) and new lineage-specific genes are generated (e.g., CKAP2, NEK5) and have become targets of rapid chromosomal diversification and positive selection (e.g., RGPDs). High-fidelity long-read sequencing has made these dynamic regions of the genome accessible for sequence-level analyses within and between primate species for the first time.
Conflict of interest statement
Competing interests E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc. The other authors declare no competing interests.
Figures




References
-
- Watson J. D. The human genome project: past, present, and future. Science 248, 44–49, (1990). - PubMed
-
- Lander E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921, (2001). - PubMed
-
- Venter J. C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous