

This is a preprint.
Leveraging Base Pair Mammalian Constraint to Understand Genetic Variation and Human Disease
- PMID: 36945512
- PMCID: PMC10028973
- DOI: 10.1101/2023.03.10.531987
Leveraging Base Pair Mammalian Constraint to Understand Genetic Variation and Human Disease
Update in
-
Leveraging base-pair mammalian constraint to understand genetic variation and human disease.Science. 2023 Apr 28;380(6643):eabn2937. doi: 10.1126/science.abn2937. Epub 2023 Apr 28. Science. 2023. PMID: 37104612 Free PMC article.
Abstract
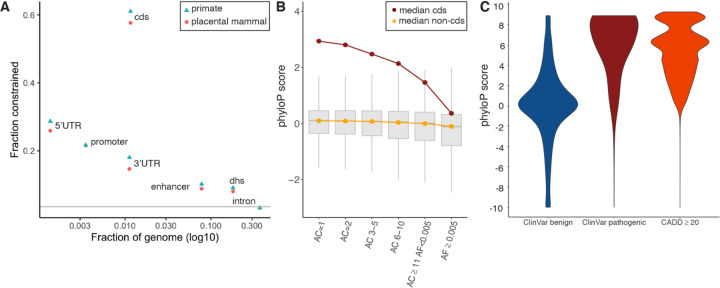
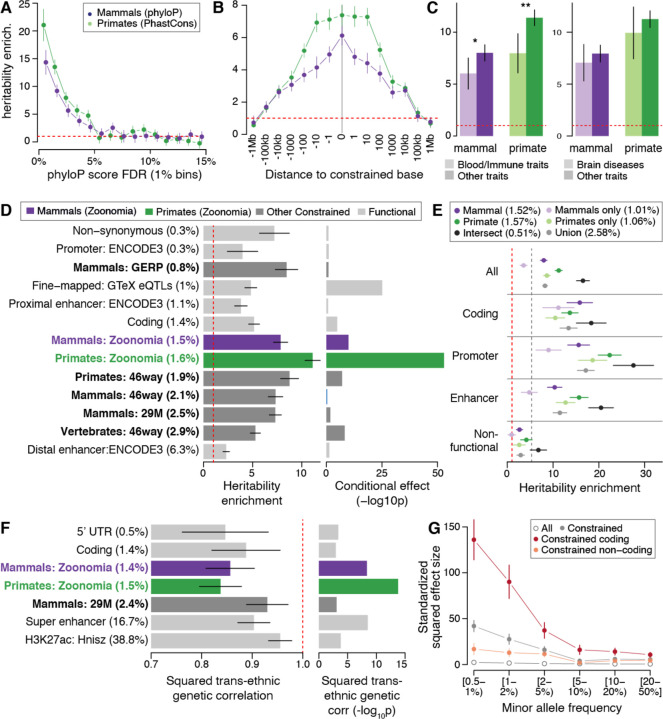
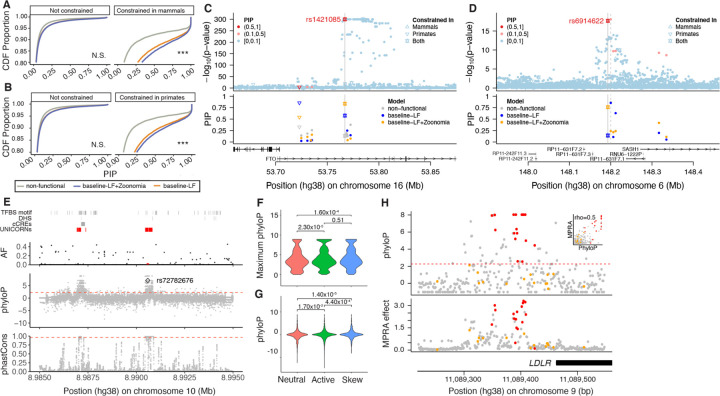
Although thousands of genomic regions have been associated with heritable human diseases, attempts to elucidate biological mechanisms are impeded by a general inability to discern which genomic positions are functionally important. Evolutionary constraint is a powerful predictor of function that is agnostic to cell type or disease mechanism. Here, single base phyloP scores from the whole genome alignment of 240 placental mammals identified 3.5% of the human genome as significantly constrained, and likely functional. We compared these scores to large-scale genome annotation, genome-wide association studies (GWAS), copy number variation, clinical genetics findings, and cancer data sets. Evolutionarily constrained positions are enriched for variants explaining common disease heritability (more than any other functional annotation). Our results improve variant annotation but also highlight that the regulatory landscape of the human genome still needs to be further explored and linked to disease.
Conflict of interest statement
Competing interests
PFS is a consultant and shareholder for Neumora.
Figures


References
-
- ENCODE Project Consortium, Moore J. E., Purcaro M. J., Pratt H. E., Epstein C. B., Shoresh N., Adrian J., Kawli T., Davis C. A., Dobin A., Kaul R., Halow J., Van Nostrand E. L., Freese P., Gorkin D. U., Shen Y., He Y., Mackiewicz M., Pauli-Behn F., Williams B. A., Mortazavi A., Keller C. A., Zhang X.-O., Elhajjajy S. I., Huey J., Dickel D. E., Snetkova V., Wei X., Wang X., Rivera-Mulia J. C., Rozowsky J., Zhang J., Chhetri S. B., Zhang J., Victorsen A., White K. P., Visel A., Yeo G. W., Burge C. B., Lécuyer E., Gilbert D. M., Dekker J., Rinn J., Mendenhall E. M., Ecker J. R., Kellis M., Klein R. J., Noble W. S., Kundaje A., Guigó R., Farnham P. J., Cherry J. M., Myers R. M., Ren B., Graveley B. R., Gerstein M. B., Pennacchio L. A., Snyder M. P., Bernstein B. E., Wold B., Hardison R. C., Gingeras T. R., Stamatoyannopoulos J. A., Weng Z., Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 583, 699–710 (2020). - PMC - PubMed
-
- Karczewski K. J., Francioli L. C., Tiao G., Cummings B. B., Alföldi J., Wang Q., Collins R. L., Laricchia K. M., Ganna A., Birnbaum D. P., Gauthier L. D., Brand H., Solomonson M., Watts N. A., Rhodes D., Singer-Berk M., England E. M., Seaby E. G., Kosmicki J. A., Walters R. K., Tashman K., Farjoun Y., Banks E., Poterba T., Wang A., Seed C., Whiffin N., Chong J. X., Samocha K. E., Pierce-Hoffman E., Zappala Z., O’Donnell-Luria A. H., Minikel E. V., Weisburd B., Lek M., Ware J. S., Vittal C., Armean I. M., Bergelson L., Cibulskis K., Connolly K. M., Covarrubias M., Donnelly S., Ferriera S., Gabriel S., Gentry J., Gupta N., Jeandet T., Kaplan D., Llanwarne C., Munshi R., Novod S., Petrillo N., Roazen D., Ruano-Rubio V., Saltzman A., Schleicher M., Soto J., Tibbetts K., Tolonen C., Wade G., Talkowski M. E., Neale B. M., Daly M. J., MacArthur D. G., The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020). - PMC - PubMed
-
- Taliun D., Harris D. N., Kessler M. D., Carlson J., Szpiech Z. A., Torres R., Taliun S. A. G., Corvelo A., Gogarten S. M., Kang H. M., Pitsillides A. N., LeFaive J., Lee S.-B., Tian X., Browning B. L., Das S., Emde A.-K., Clarke W. E., Loesch D. P., Shetty A. C., Blackwell T. W., Smith A. V., Wong Q., Liu X., Conomos M. P., Bobo D. M., Aguet F., Albert C., Alonso A., Ardlie K. G., Arking D. E., Aslibekyan S., Auer P. L., Barnard J., Barr R. G., Barwick L., Becker L. C., Beer R. L., Benjamin E. J., Bielak L. F., Blangero J., Boehnke M., Bowden D. W., Brody J. A., Burchard E. G., Cade B. E., Casella J. F., Chalazan B., Chasman D. I., Chen Y.-D. I., Cho M. H., Choi S. H., Chung M. K., Clish C. B., Correa A., Curran J. E., Custer B., Darbar D., Daya M., de Andrade M., DeMeo D. L., Dutcher S. K., Ellinor P. T., Emery L. S., Eng C., Fatkin D., Fingerlin T., Forer L., Fornage M., Franceschini N., Fuchsberger C., Fullerton S. M., Germer S., Gladwin M. T., Gottlieb D. J., Guo X., Hall M. E., He J., Heard-Costa N. L., Heckbert S. R., Irvin M. R., Johnsen J. M., Johnson A. D., Kaplan R., Kardia S. L. R., Kelly T., Kelly S., Kenny E. E., Kiel D. P., Klemmer R., Konkle B. A., Kooperberg C., Köttgen A., Lange L. A., Lasky-Su J., Levy D., Lin X., Lin K.-H., Liu C., Loos R. J. F., Garman L., Gerszten R., Lubitz S. A., Lunetta K. L., Mak A. C. Y., Manichaikul A., Manning A. K., Mathias R. A., McManus D. D., McGarvey S. T., Meigs J. B., Meyers D. A., Mikulla J. L., Minear M. A., Mitchell B. D., Mohanty S., Montasser M. E., Montgomery C., Morrison A. C., Murabito J. M., Natale A., Natarajan P., Nelson S. C., North K. E., O’Connell J. R., Palmer N. D., Pankratz N., Peloso G. M., Peyser P. A., Pleiness J., Post W. S., Psaty B. M., Rao D. C., Redline S., Reiner A. P., Roden D., Rotter J. I., Ruczinski I., Sarnowski C., Schoenherr S., Schwartz D. A., Seo J.-S., Seshadri S., Sheehan V. A., Sheu W. H., Shoemaker M. B., Smith N. L., Smith J. A., Sotoodehnia N., Stilp A. M., Tang W., Taylor K. D., Telen M., Thornton T. A., Tracy R. P., Van Den Berg D. J., Vasan R. S., Viaud-Martinez K. A., Vrieze S., Weeks D. E., Weir B. S., Weiss S. T., Weng L.-C., Willer C. J., Zhang Y., Zhao X., Arnett D. K., Ashley-Koch A. E., Barnes K. C., Boerwinkle E., Gabriel S., Gibbs R., Rice K. M., Rich S. S., Silverman E. K., Qasba P., Gan W., NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, Papanicolaou G. J., Nickerson D. A., Browning S. R., Zody M. C., Zöllner S., Wilson J. G., Cupples L. A., Laurie C. C., Jaquish C. E., Hernandez R. D., O’Connor T. D., Abecasis G. R., Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 590, 290–299 (2021). - PMC - PubMed
-
- Cooper G. M., Shendure J., Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nature Reviews Genetics. 12 (2011), pp. 628–640. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources