

This is a preprint.
Combining pairwise structural similarity and deep learning interface contact prediction to estimate protein complex model accuracy in CASP15
- PMID: 36945536
- PMCID: PMC10028888
- DOI: 10.1101/2023.03.08.531814
Combining pairwise structural similarity and deep learning interface contact prediction to estimate protein complex model accuracy in CASP15
Update in
-
Combining pairwise structural similarity and deep learning interface contact prediction to estimate protein complex model accuracy in CASP15.Proteins. 2023 Dec;91(12):1889-1902. doi: 10.1002/prot.26542. Epub 2023 Jun 26. Proteins. 2023. PMID: 37357816 Free PMC article.
Abstract
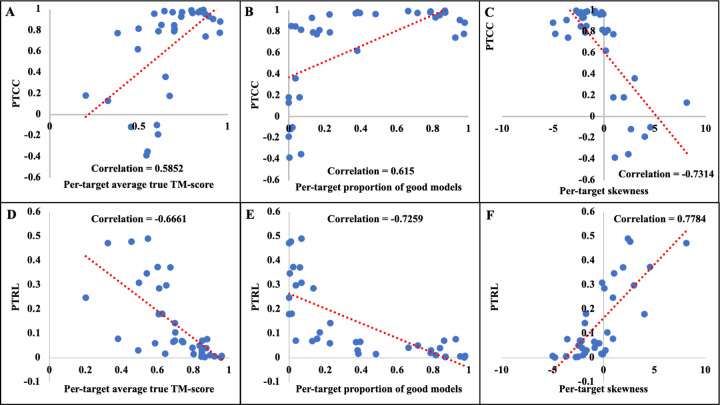
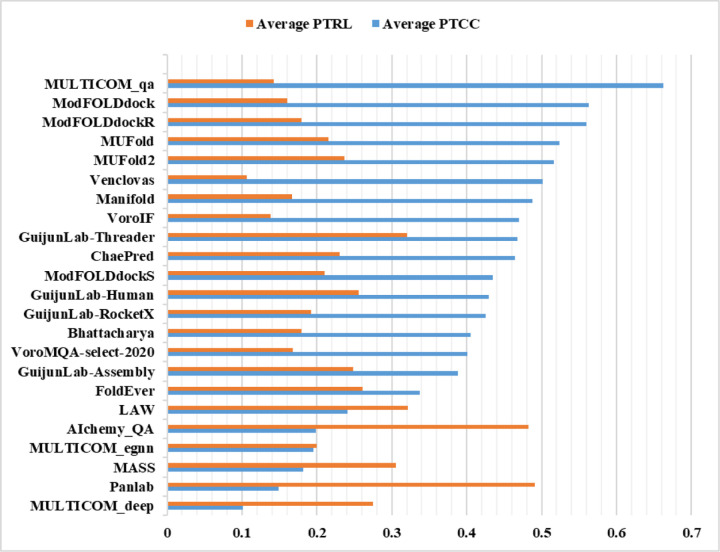
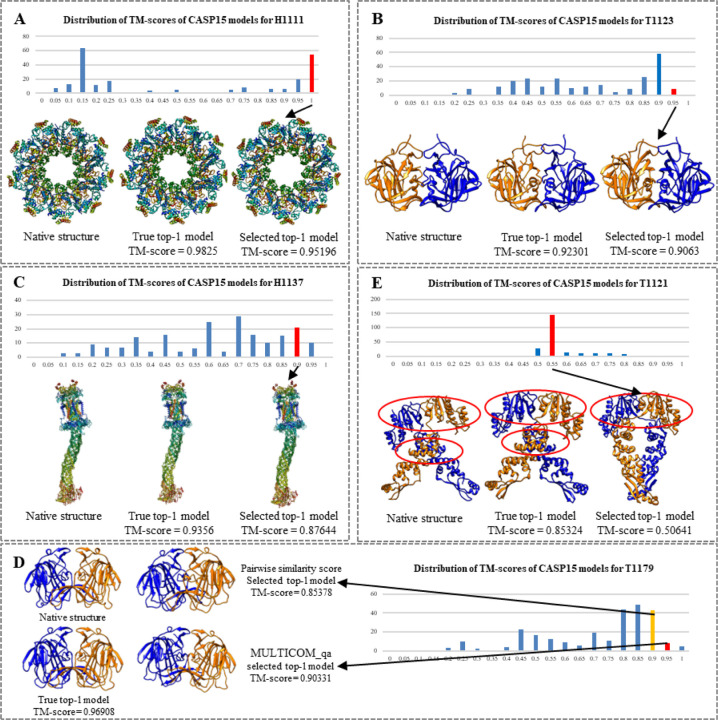
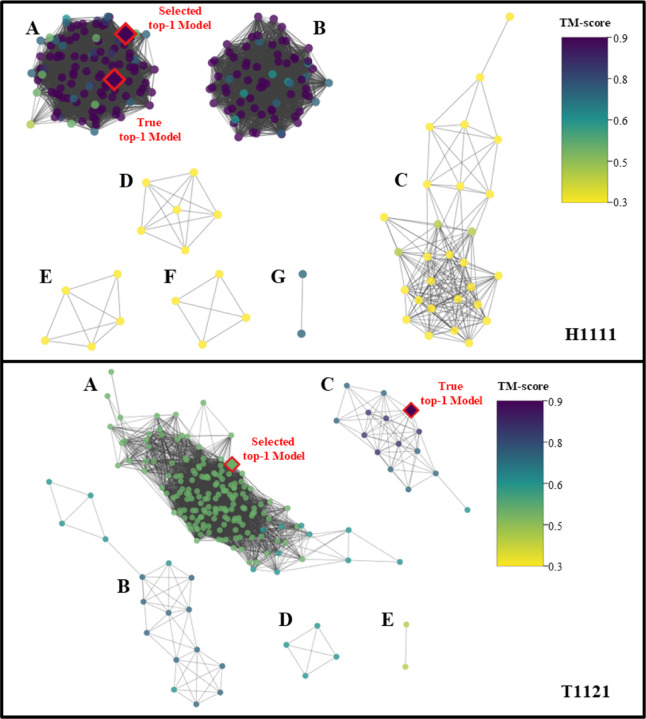
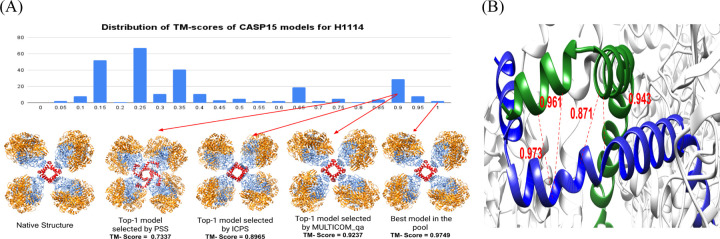
Estimating the accuracy of quaternary structural models of protein complexes and assemblies (EMA) is important for predicting quaternary structures and applying them to studying protein function and interaction. The pairwise similarity between structural models is proven useful for estimating the quality of protein tertiary structural models, but it has been rarely applied to predicting the quality of quaternary structural models. Moreover, the pairwise similarity approach often fails when many structural models are of low quality and similar to each other. To address the gap, we developed a hybrid method (MULTICOM_qa) combining a pairwise similarity score (PSS) and an interface contact probability score (ICPS) based on the deep learning inter-chain contact prediction for estimating protein complex model accuracy. It blindly participated in the 15th Critical Assessment of Techniques for Protein Structure Prediction (CASP15) in 2022 and ranked first out of 24 predictors in estimating the global accuracy of assembly models. The average per-target correlation coefficient between the model quality scores predicted by MULTICOM_qa and the true quality scores of the models of CASP15 assembly targets is 0.66. The average per-target ranking loss in using the predicted quality scores to rank the models is 0.14. It was able to select good models for most targets. Moreover, several key factors (i.e., target difficulty, model sampling difficulty, skewness of model quality, and similarity between good/bad models) for EMA are identified and analayzed. The results demonstrate that combining the multi-model method (PSS) with the complementary single-model method (ICPS) is a promising approach to EMA. The source code of MULTICOM_qa is available at https://github.com/BioinfoMachineLearning/MULTICOM_qa .
Figures
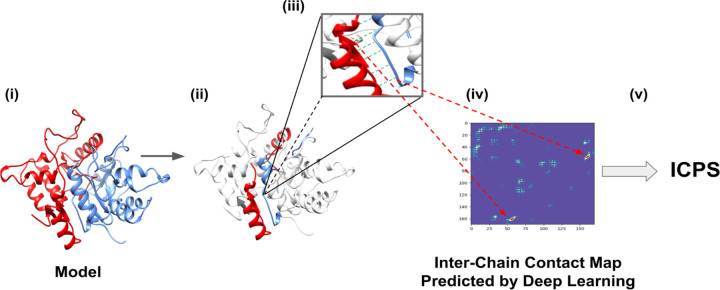
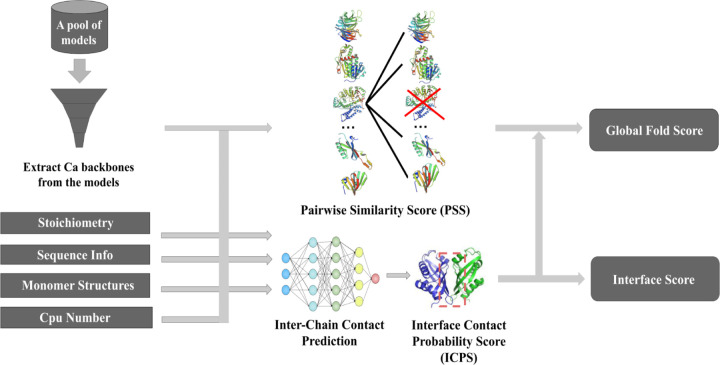
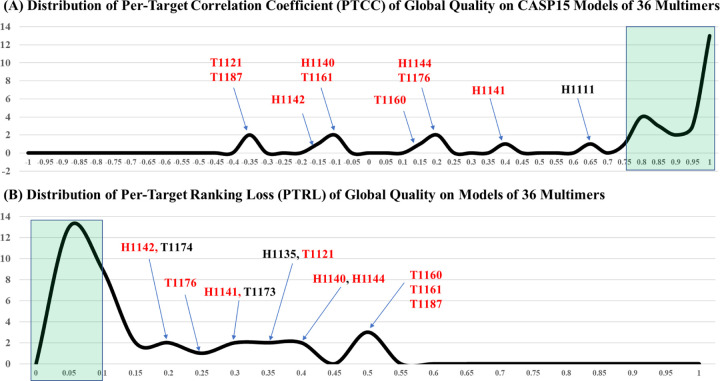
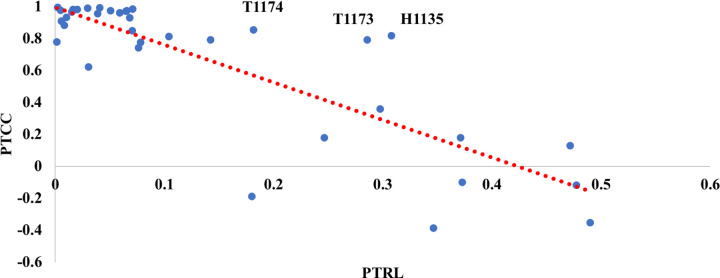
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous