

This is a preprint.
Isogenic hiPSC models of Turner syndrome development reveal shared roles of inactive X and Y in the human cranial neural crest network
- PMID: 36945647
- PMCID: PMC10028916
- DOI: 10.1101/2023.03.08.531747
Isogenic hiPSC models of Turner syndrome development reveal shared roles of inactive X and Y in the human cranial neural crest network
Update in
-
Isogenic hiPSC models of Turner syndrome development reveal shared roles of inactive X and Y in the human cranial neural crest network.Am J Hum Genet. 2025 Mar 6;112(3):615-629. doi: 10.1016/j.ajhg.2025.01.013. Epub 2025 Feb 7. Am J Hum Genet. 2025. PMID: 39922196 Free PMC article.
Abstract
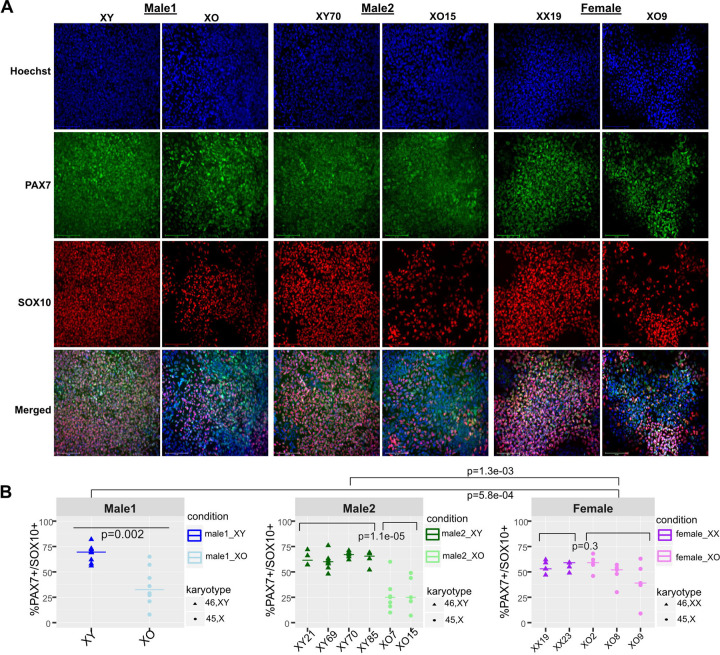
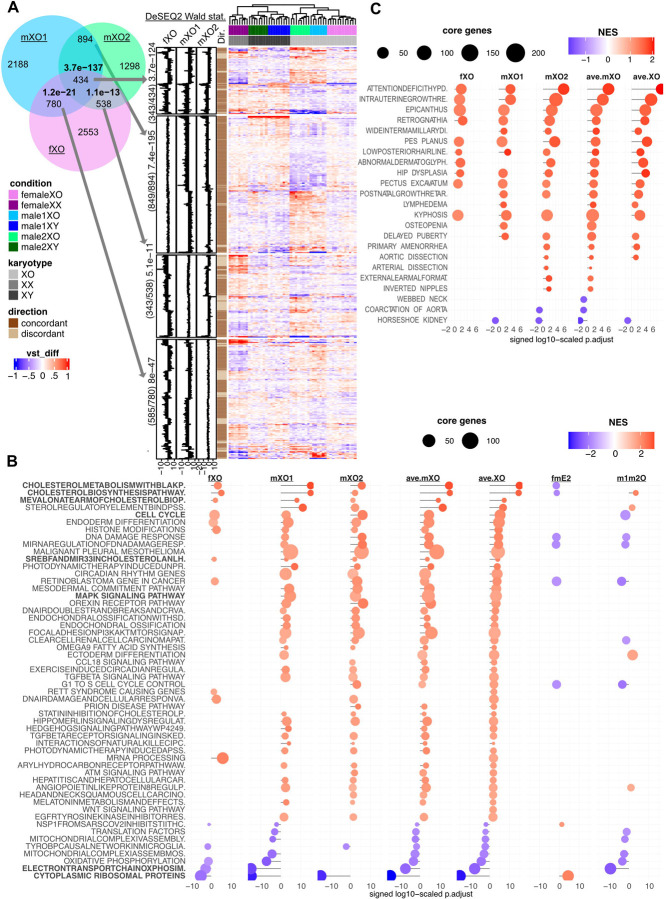
Modeling the developmental etiology of viable human aneuploidy can be challenging in rodents due to syntenic boundaries, or primate-specific biology. In humans, monosomy-X (45,X) causes Turner syndrome (TS), altering craniofacial, skeletal, endocrine, and cardiovascular development, which in contrast remain unaffected in 39,X-mice. To learn how human monosomy-X may impact early embryonic development, we turned to human 45,X and isogenic euploid induced pluripotent stem cells (hiPSCs) from male and female mosaic donors. Because neural crest (NC) derived cell types are hypothesized to underpin craniofacial and cardiovascular changes in TS, we performed a highly-powered differential expression study on hiPSC-derived anterior neural crest cells (NCCs). Across three independent isogenic panels, 45,X NCCs show impaired acquisition of PAX7+SOX10+ markers, and disrupted expression of other NCC-specific genes, relative to their isogenic euploid controls. In particular, 45,X NCCs increase cholesterol biosynthesis genes while reducing transcripts that feature 5' terminal oligopyrimidine (TOP) motifs, including those of ribosomal protein and nuclear-encoded mitochondrial genes. Such metabolic pathways are also over-represented in weighted co-expression gene modules that are preserved in monogenic neurocristopathy. Importantly, these gene modules are also significantly enriched in 28% of all TS-associated terms of the human phenotype ontology. Our analysis identifies specific sex-linked genes that are expressed from two copies in euploid males and females alike and qualify as candidate haploinsufficient drivers of TS phenotypes in NC-derived lineages. This study demonstrates that isogenic hiPSC-derived NCC panels representing monosomy-X can serve as a powerful model of early NC development in TS and inform new hypotheses towards its etiology.
Figures


References
-
- Hook E. B. & Warburton D. The distribution of chromosomal genotypes associated with Turner’s syndrome: livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. Hum. Genet. 64, 24–7 (1983). - PubMed
-
- Hook E. B. & Warburton D. Turner syndrome revisited: review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum. Genet. 133, 417–24 (2014). - PubMed
-
- Gravholt C. H., Viuff M. H., Brun S., Stochholm K. & Andersen N. H. Turner syndrome: mechanisms and management. Nat. Rev. Endocrinol. 15, 601–614 (2019). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources