

Cell-Autonomous Cxcl1 Sustains Tolerogenic Circuitries and Stromal Inflammation via Neutrophil-Derived TNF in Pancreatic Cancer
- PMID: 36946782
- PMCID: PMC10259764
- DOI: 10.1158/2159-8290.CD-22-1046
Cell-Autonomous Cxcl1 Sustains Tolerogenic Circuitries and Stromal Inflammation via Neutrophil-Derived TNF in Pancreatic Cancer
Abstract
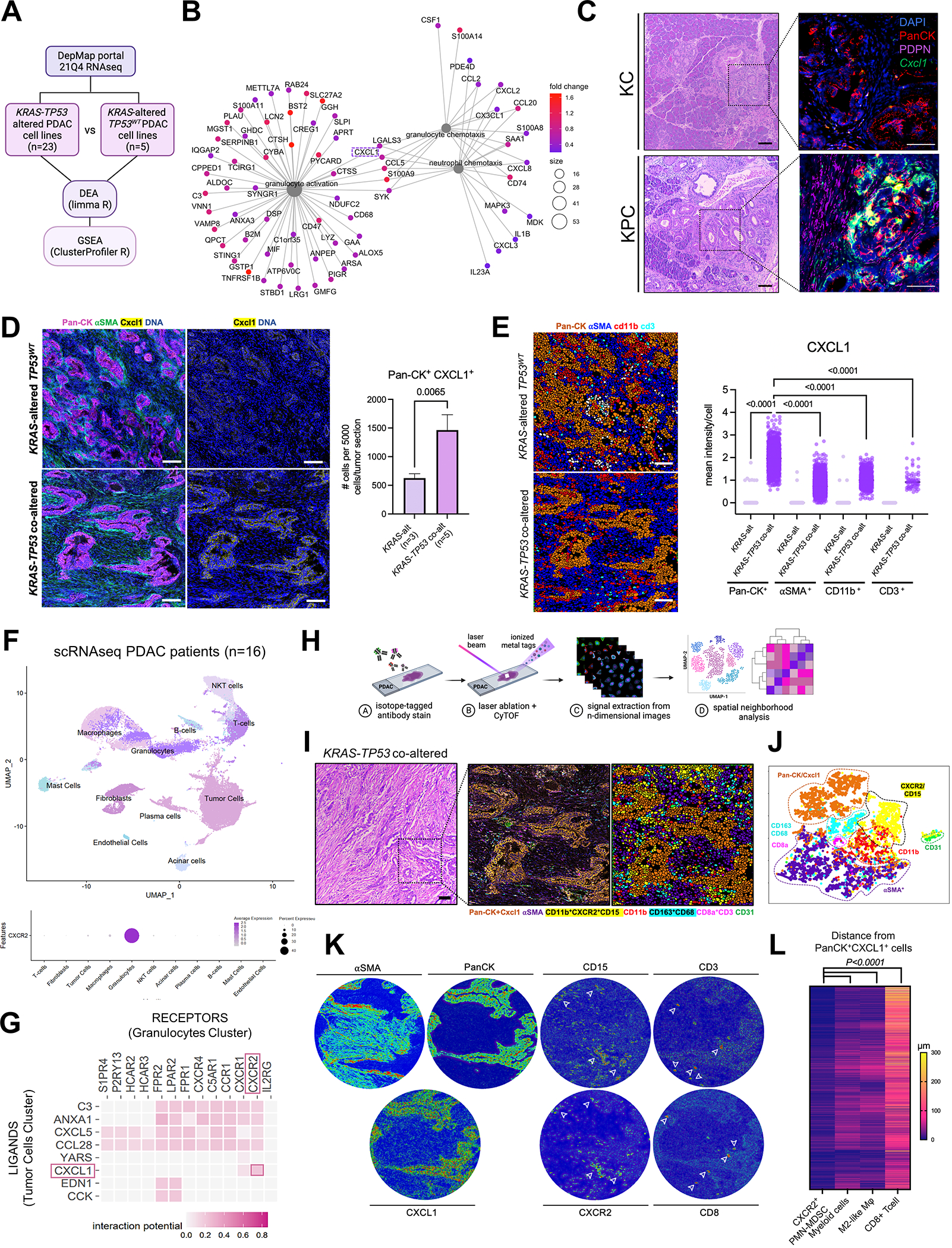
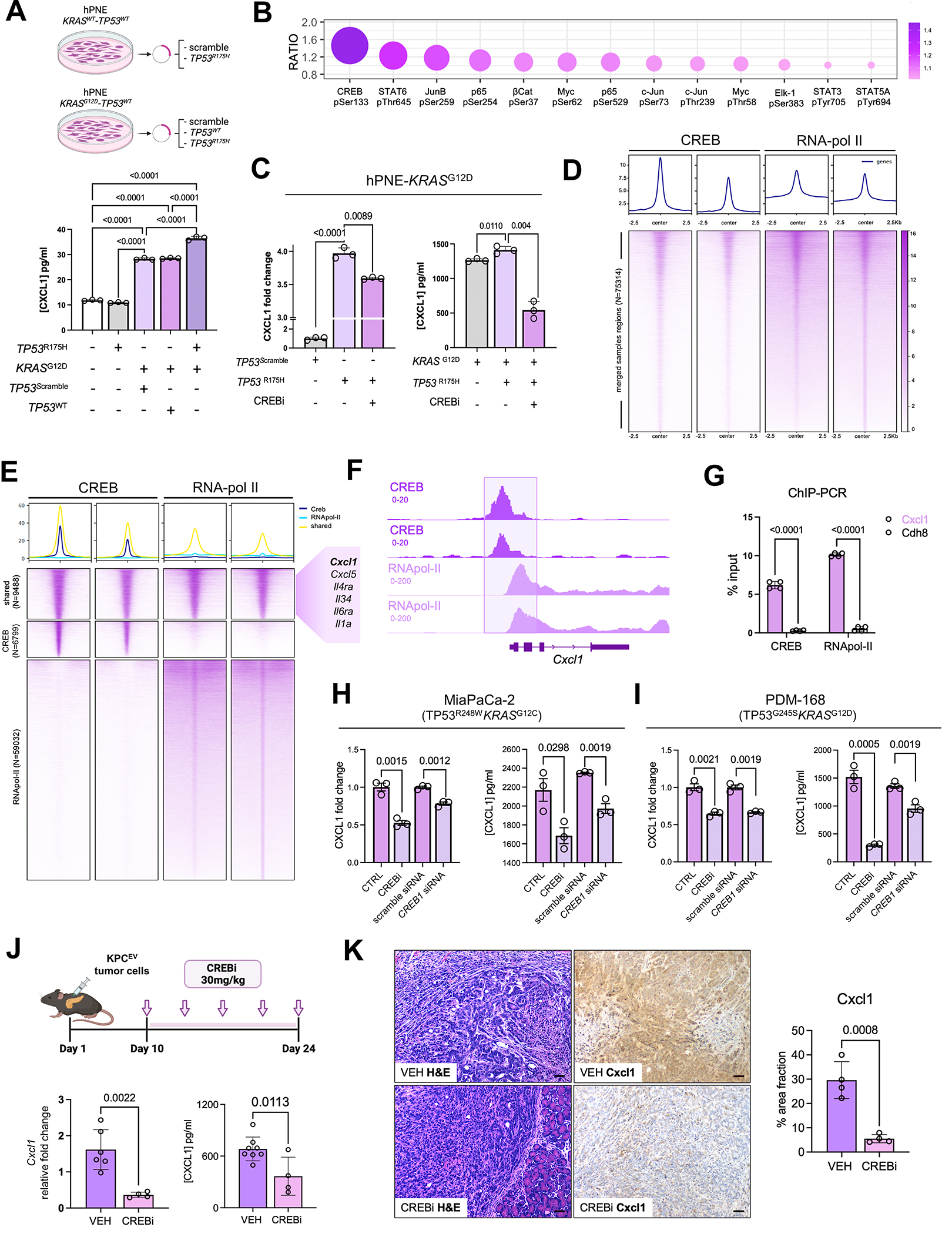
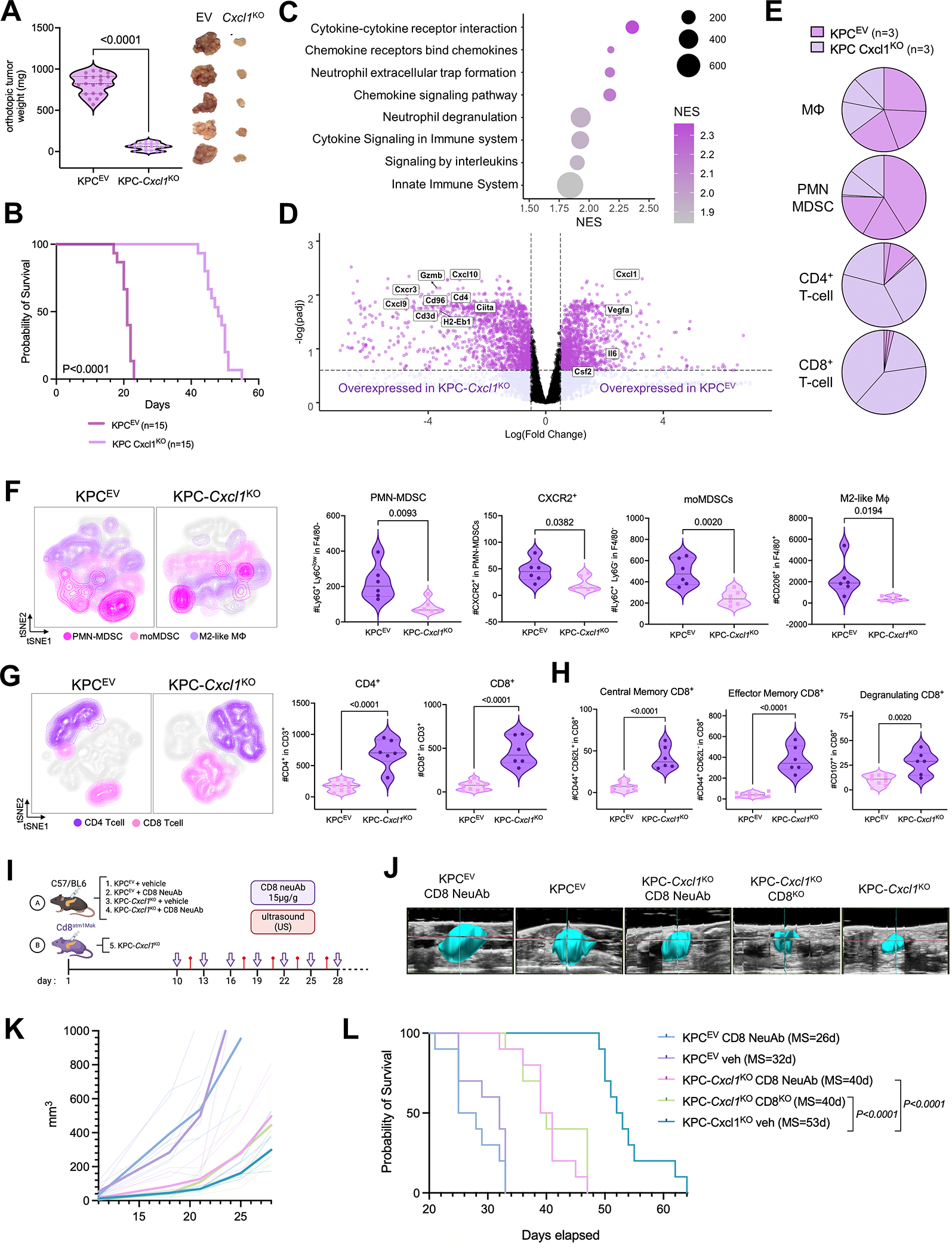
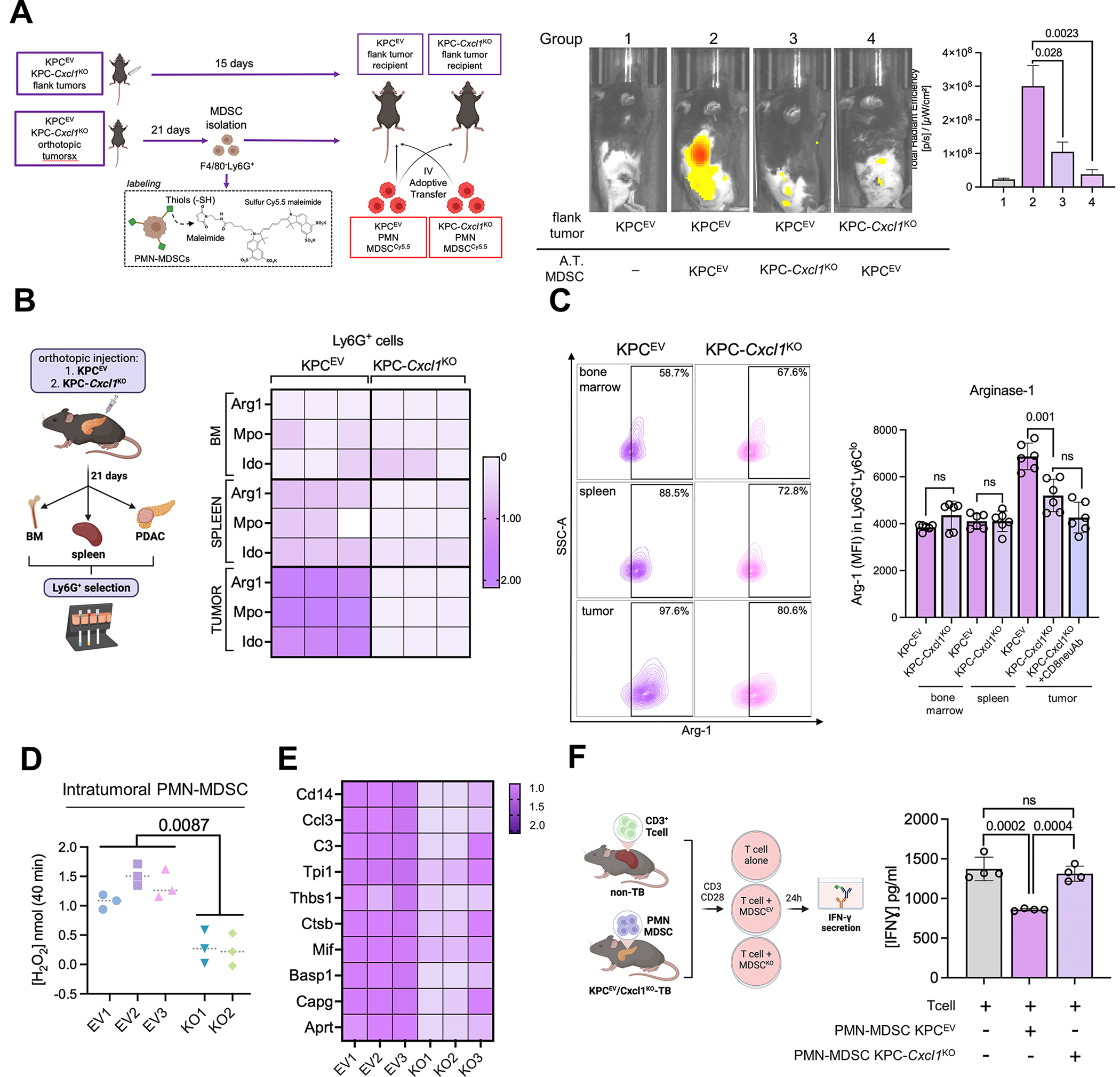
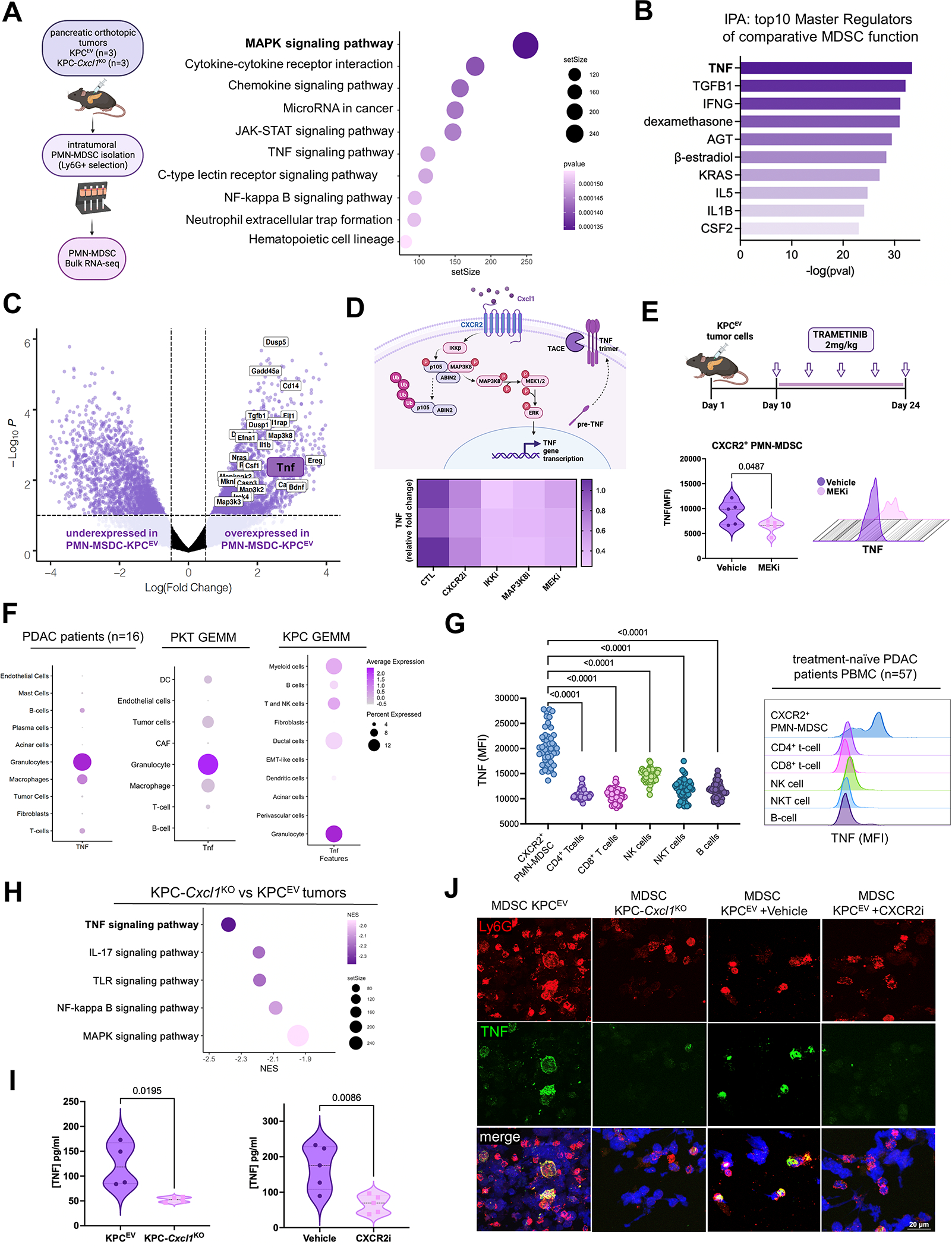
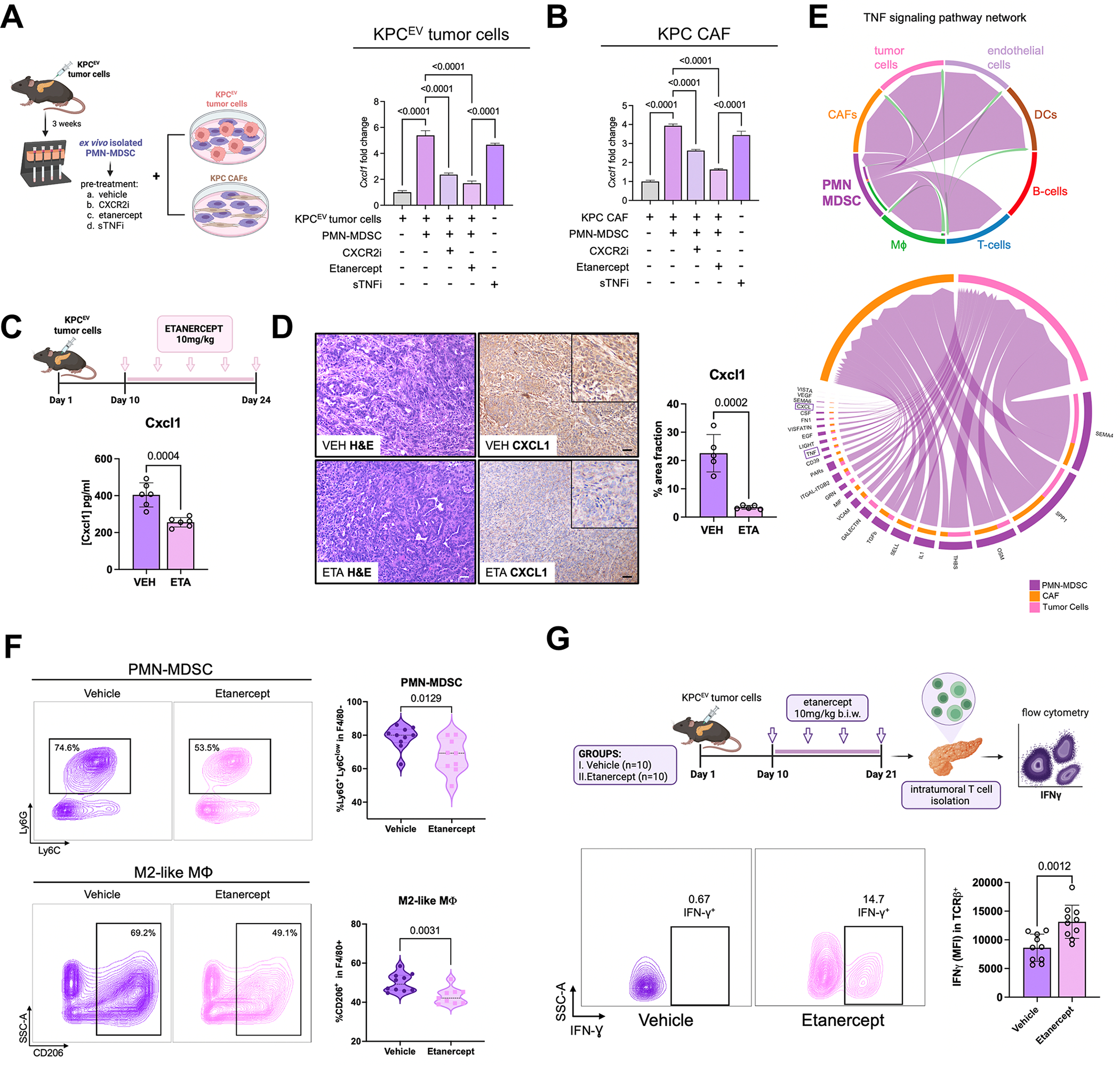
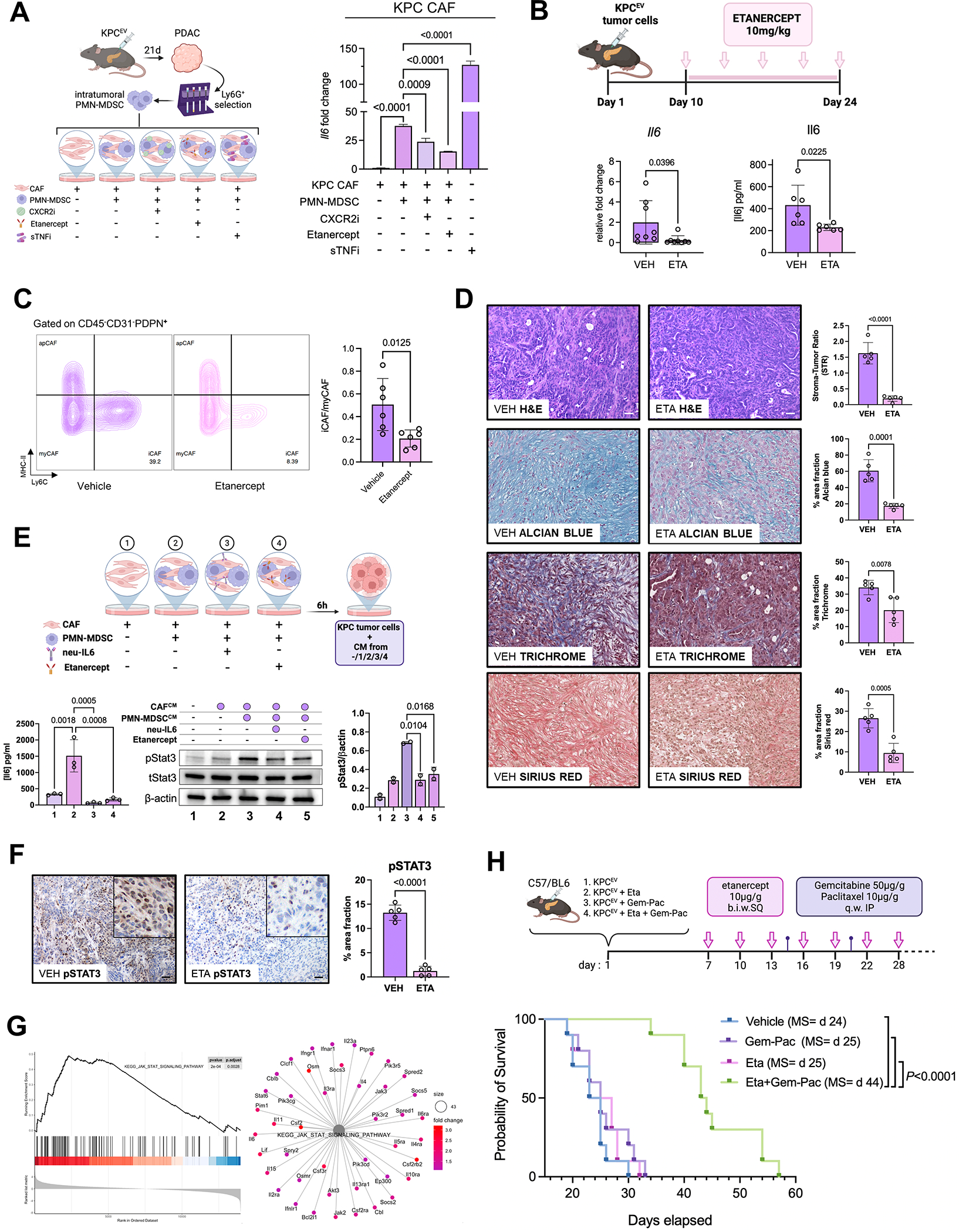
We have shown that KRAS-TP53 genomic coalteration is associated with immune-excluded microenvironments, chemoresistance, and poor survival in pancreatic ductal adenocarcinoma (PDAC) patients. By treating KRAS-TP53 cooperativity as a model for high-risk biology, we now identify cell-autonomous Cxcl1 as a key mediator of spatial T-cell restriction via interactions with CXCR2+ neutrophilic myeloid-derived suppressor cells in human PDAC using imaging mass cytometry. Silencing of cell-intrinsic Cxcl1 in LSL-KrasG12D/+;Trp53R172H/+;Pdx-1Cre/+(KPC) cells reprograms the trafficking and functional dynamics of neutrophils to overcome T-cell exclusion and controls tumor growth in a T cell-dependent manner. Mechanistically, neutrophil-derived TNF is a central regulator of this immunologic rewiring, instigating feed-forward Cxcl1 overproduction from tumor cells and cancer-associated fibroblasts (CAF), T-cell dysfunction, and inflammatory CAF polarization via transmembrane TNF-TNFR2 interactions. TNFR2 inhibition disrupts this circuitry and improves sensitivity to chemotherapy in vivo. Our results uncover cancer cell-neutrophil cross-talk in which context-dependent TNF signaling amplifies stromal inflammation and immune tolerance to promote therapeutic resistance in PDAC.
Significance: By decoding connections between high-risk tumor genotypes, cell-autonomous inflammatory programs, and myeloid-enriched/T cell-excluded contexts, we identify a novel role for neutrophil-derived TNF in sustaining immunosuppression and stromal inflammation in pancreatic tumor microenvironments. This work offers a conceptual framework by which targeting context-dependent TNF signaling may overcome hallmarks of chemoresistance in pancreatic cancer. This article is highlighted in the In This Issue feature, p. 1275.
©2023 American Association for Cancer Research.
Conflict of interest statement
Figures
Comment in
-
Neutrophil-sourced TNF in cancer: deciphering an intricate orchestrator of immunosuppressive communication in the tumor microenvironment.Signal Transduct Target Ther. 2023 Jul 17;8(1):272. doi: 10.1038/s41392-023-01530-4. Signal Transduct Target Ther. 2023. PMID: 37455286 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
