

Sexual behavior shapes male genitourinary microbiome composition
- PMID: 36948151
- PMCID: PMC10040456
- DOI: 10.1016/j.xcrm.2023.100981
Sexual behavior shapes male genitourinary microbiome composition
Abstract
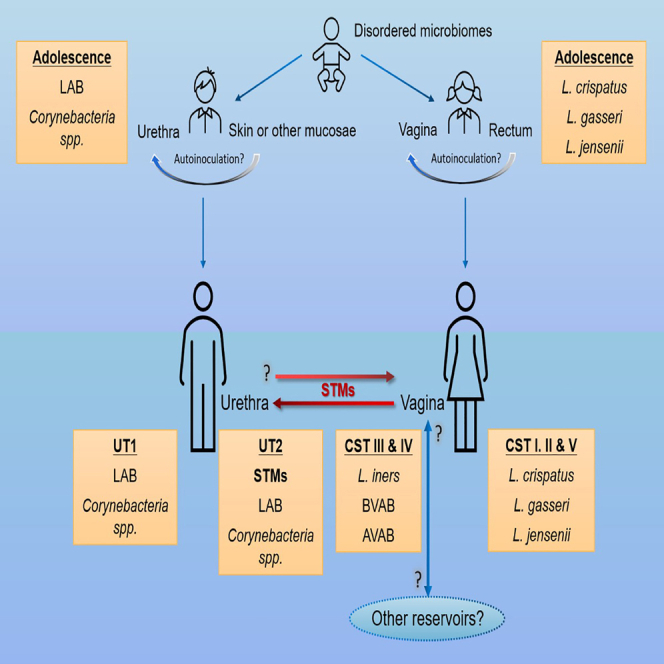
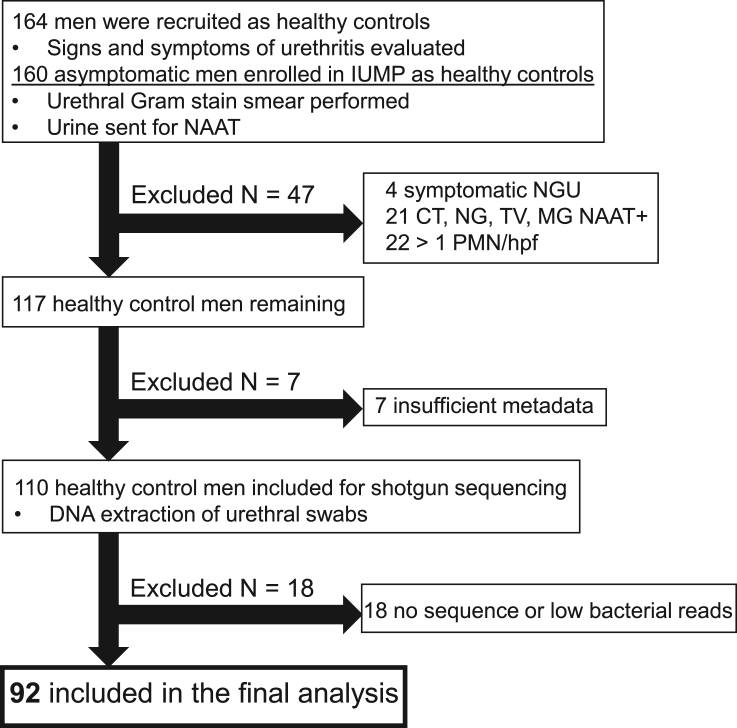
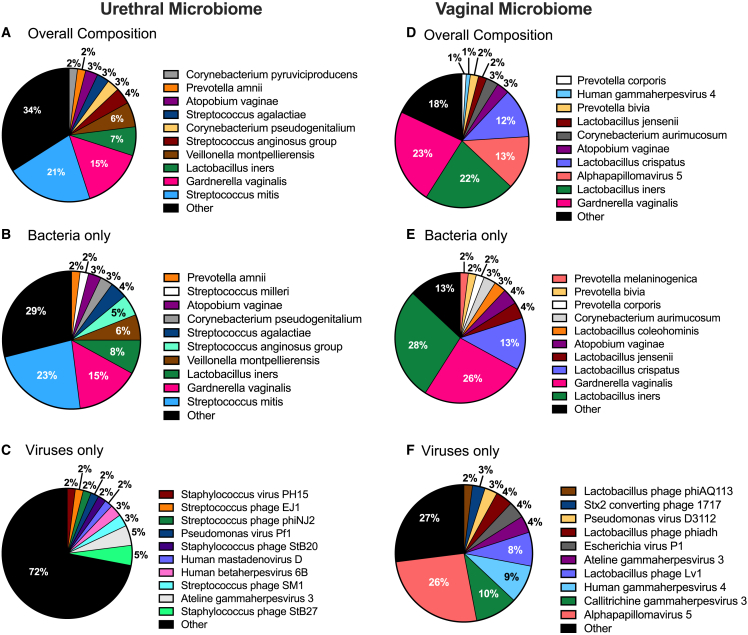
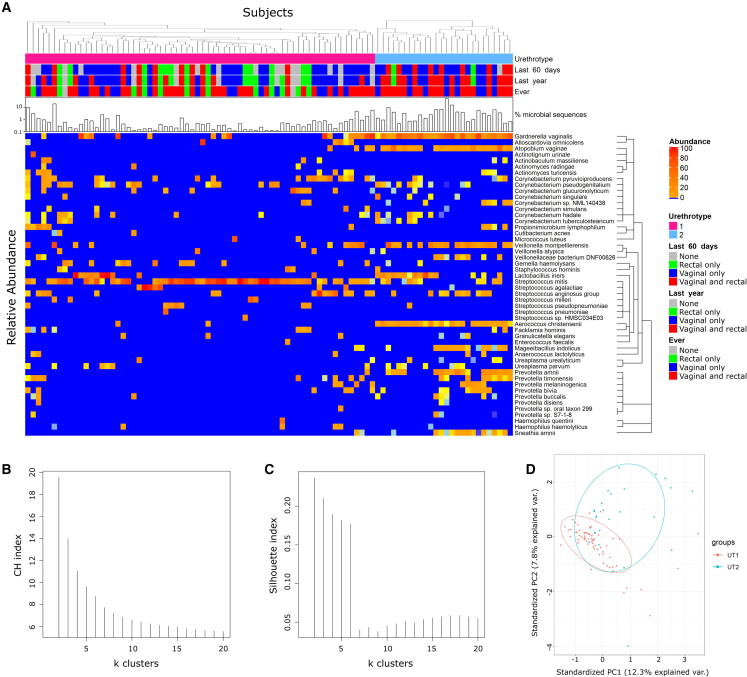
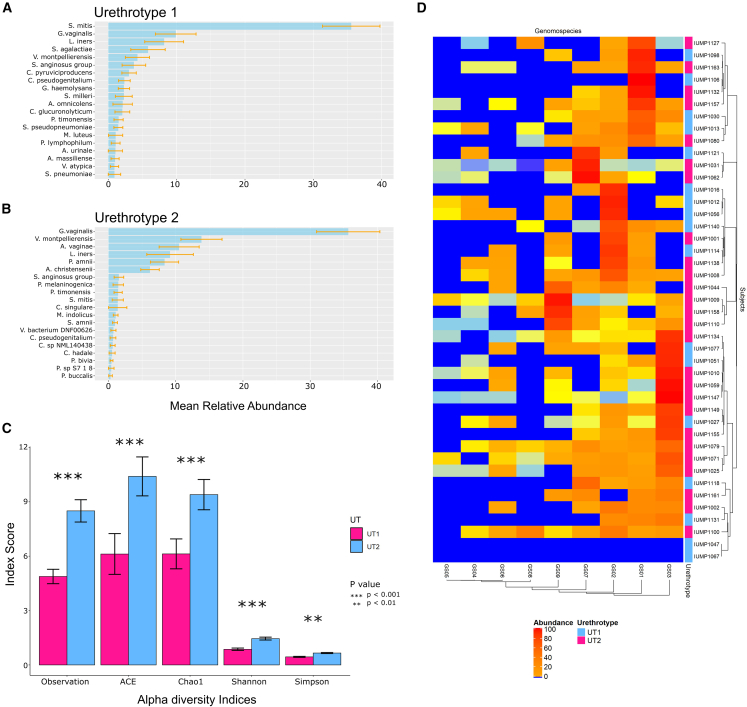
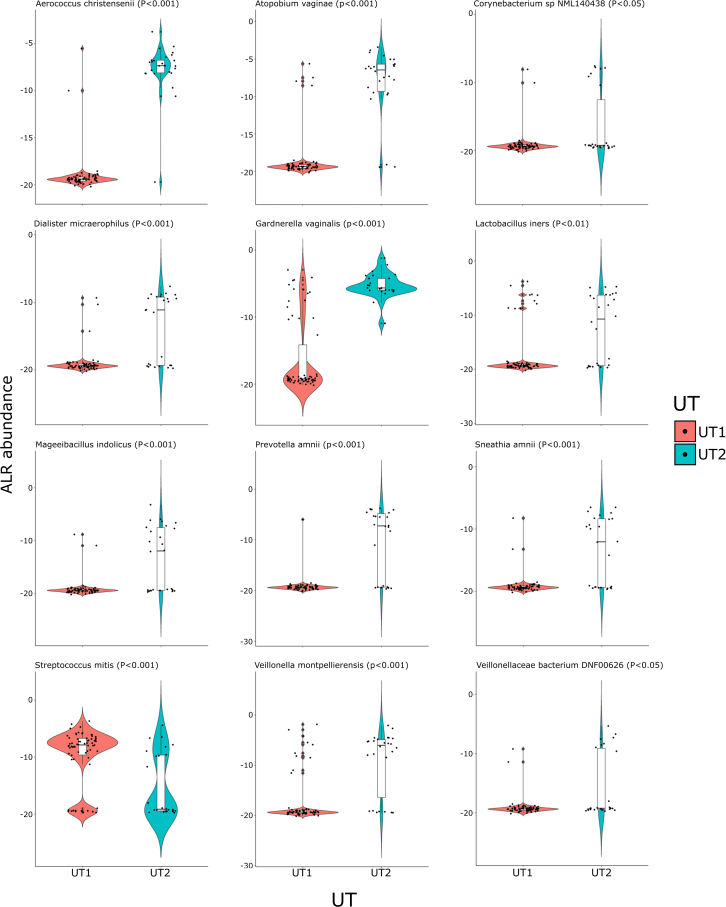
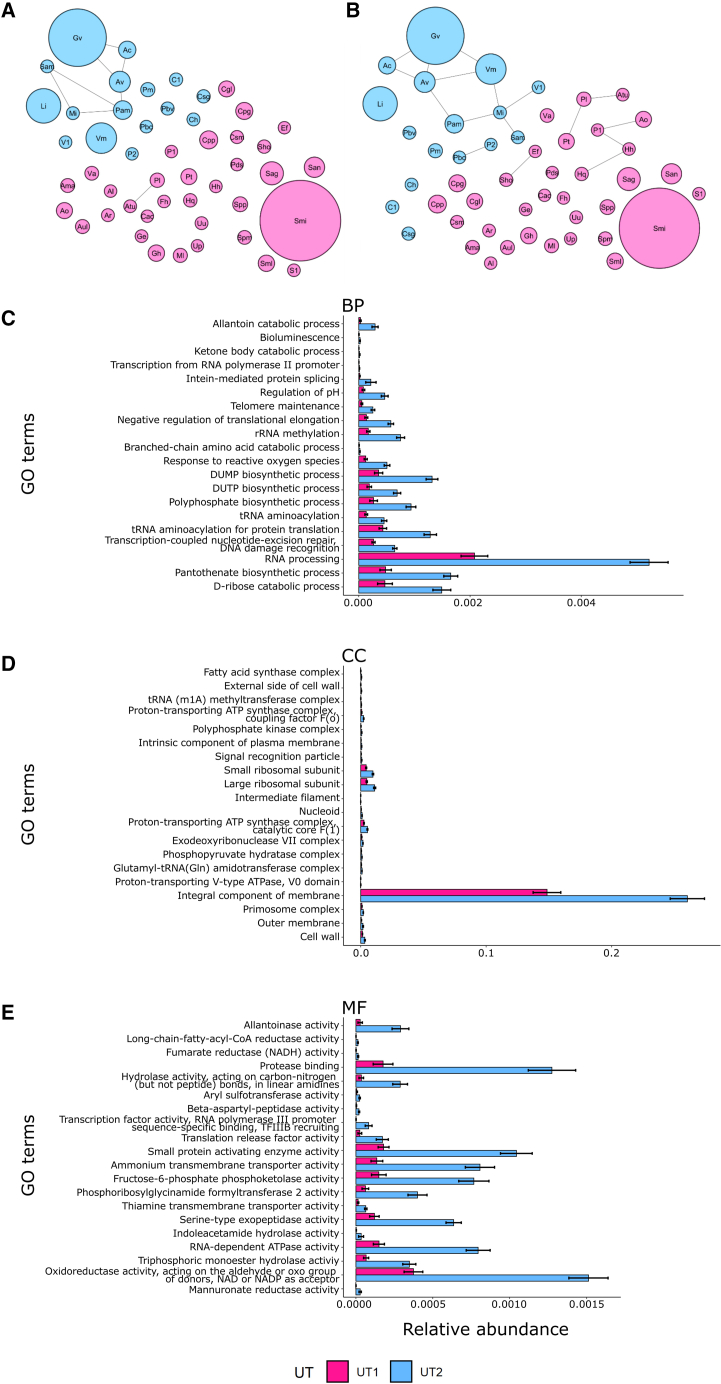
The origin, composition, and significance of the distal male urethral microbiome are unclear, but vaginal microbiome dysbiosis is linked to new sex partners and several urogynecological syndromes. We characterized 110 urethral specimens from men without urethral symptoms, infections, or inflammation using shotgun metagenomics. Most urethral specimens contain characteristic lactic acid bacteria and Corynebacterium spp. In contrast, several bacteria associated with vaginal dysbiosis were present only in specimens from men who reported vaginal intercourse. Sexual behavior, but not other evaluated behavioral, demographic, or clinical variables, strongly associated with inter-specimen variance in urethral microbiome composition. Thus, the male urethra supports a simple core microbiome that is established independent of sexual exposures but can be re-shaped by vaginal sex. Overall, the results suggest that urogenital microbiology and sexual behavior are inexorably intertwined, and show that the male urethra harbors female urogenital pathobionts.
Keywords: development; idiopathic; microbiome; microbiota; sexually transmitted infection; urethra; urethritis; urobiome.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Y.X., X.G., T.A.B., B.V.D.P., N.G., L.J.F., J.D.F., and Q.D. have no conflicts of interest. S.J.J. has received honoraria and consulting fees from Hologic, Inc. E.T., J.A.W., and. D.E.N. retain the patent for the US_NmUC diagnostic assay used in this manuscript. C.A.M. is a consultant for Lupin Pharmaceuticals, BioFire Diagnostics, Cepheid, and PhagoMed. She has also received research funding support from Lupin Pharmaceuticals, Abbott Molecular, and Gilead as well as speaker honoraria from Abbott Molecular, Cepheid, Roche Diagnostics, and Becton Dickinson.
Figures
Comment in
-
Re: Sexual Behavior Shapes Male Genitourinary Microbiome Composition.Eur Urol. 2023 Dec;84(6):598-599. doi: 10.1016/j.eururo.2023.06.028. Epub 2023 Jul 20. Eur Urol. 2023. PMID: 37481417 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
