

Assessment of community efforts to advance network-based prediction of protein-protein interactions
- PMID: 36949045
- PMCID: PMC10033937
- DOI: 10.1038/s41467-023-37079-7
Assessment of community efforts to advance network-based prediction of protein-protein interactions
Abstract
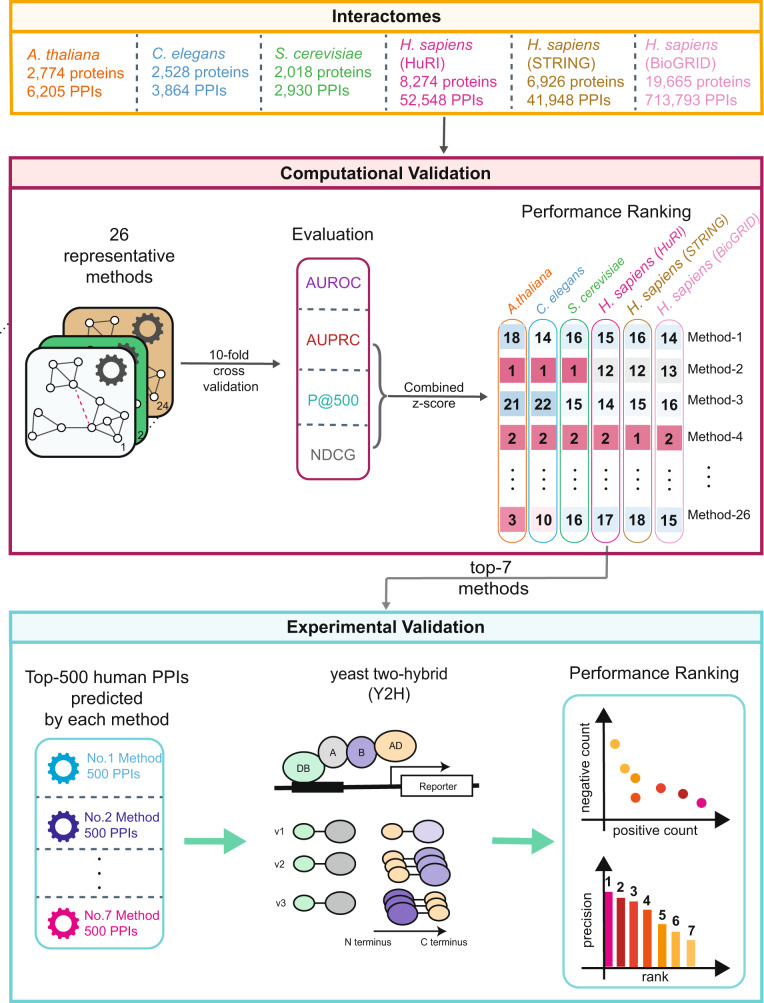
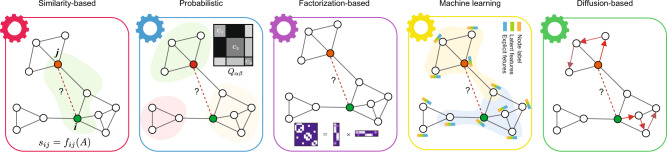
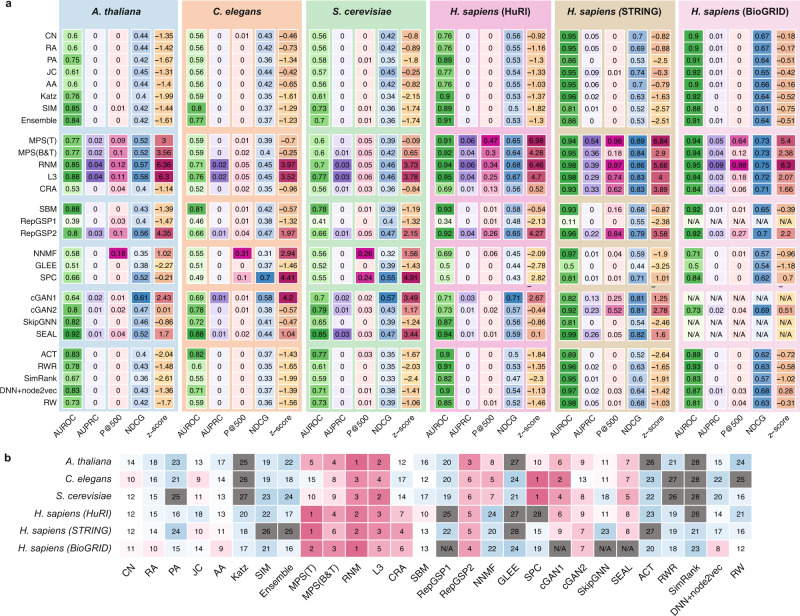
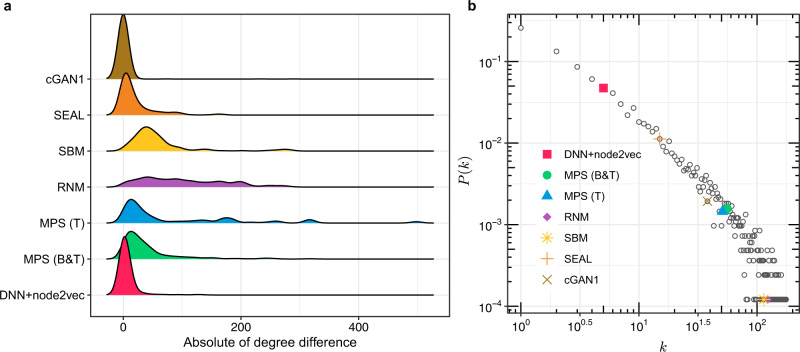
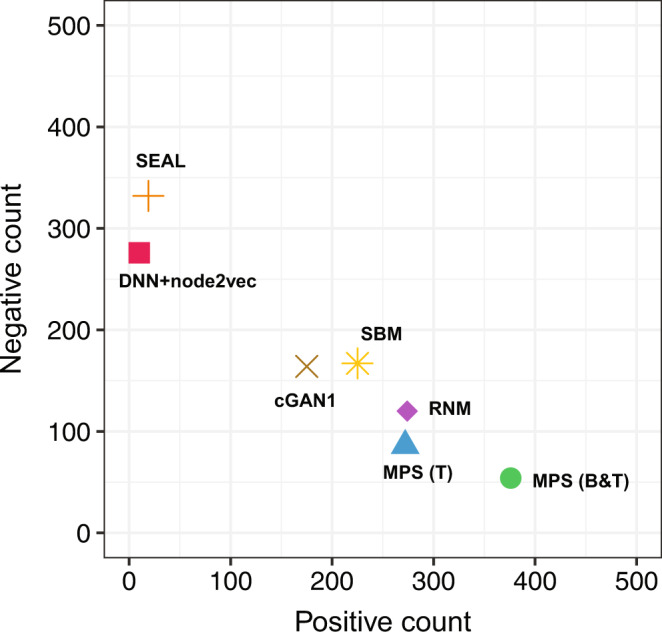
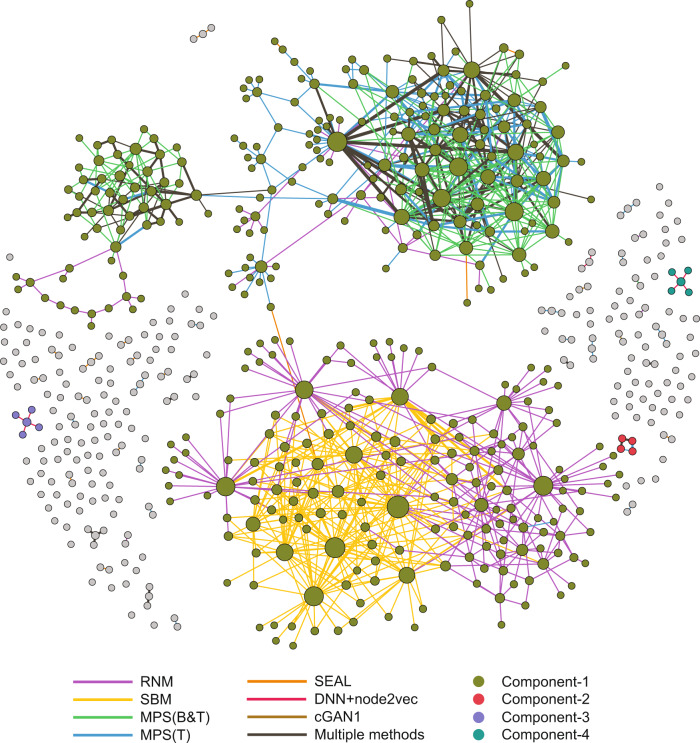
Comprehensive understanding of the human protein-protein interaction (PPI) network, aka the human interactome, can provide important insights into the molecular mechanisms of complex biological processes and diseases. Despite the remarkable experimental efforts undertaken to date to determine the structure of the human interactome, many PPIs remain unmapped. Computational approaches, especially network-based methods, can facilitate the identification of previously uncharacterized PPIs. Many such methods have been proposed. Yet, a systematic evaluation of existing network-based methods in predicting PPIs is still lacking. Here, we report community efforts initiated by the International Network Medicine Consortium to benchmark the ability of 26 representative network-based methods to predict PPIs across six different interactomes of four different organisms: A. thaliana, C. elegans, S. cerevisiae, and H. sapiens. Through extensive computational and experimental validations, we found that advanced similarity-based methods, which leverage the underlying network characteristics of PPIs, show superior performance over other general link prediction methods in the interactomes we considered.
© 2023. The Author(s).
Conflict of interest statement
PF is the founder and CEO of Pharmahungary Group, a group of R&D companies. EKS has received institutional grant support from Bayer and GlaxoSimthKline. A-LB is co-scientific founder of and is supported by Scipher Medicine, Inc., which applies network medicine strategies to biomarker development and personalized drug selection, and is the founder of Naring Inc., which applies data science to health and nutrition. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
- U01 HG007690/HG/NHGRI NIH HHS/United States
- R01 HL155107/HL/NHLBI NIH HHS/United States
- U19 AI095219/AI/NIAID NIH HHS/United States
- RF1 AG067744/AG/NIA NIH HHS/United States
- R01 GM130885/GM/NIGMS NIH HHS/United States
- R01 AI141529/AI/NIAID NIH HHS/United States
- R01 HD093761/HD/NICHD NIH HHS/United States
- U54 HL119145/HL/NHLBI NIH HHS/United States
- R01 HL155096/HL/NHLBI NIH HHS/United States
- U01 HL089856/HL/NHLBI NIH HHS/United States
- P01 HL132825/HL/NHLBI NIH HHS/United States
- UH3 OD023268/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
