

Integrated analysis of genomic and transcriptomic data for the discovery of splice-associated variants in cancer
- PMID: 36949070
- PMCID: PMC10033906
- DOI: 10.1038/s41467-023-37266-6
Integrated analysis of genomic and transcriptomic data for the discovery of splice-associated variants in cancer
Abstract
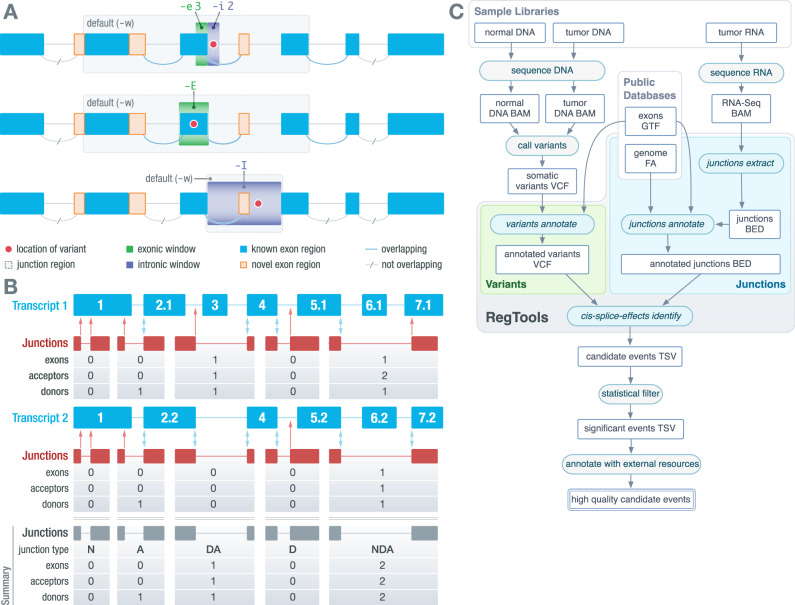
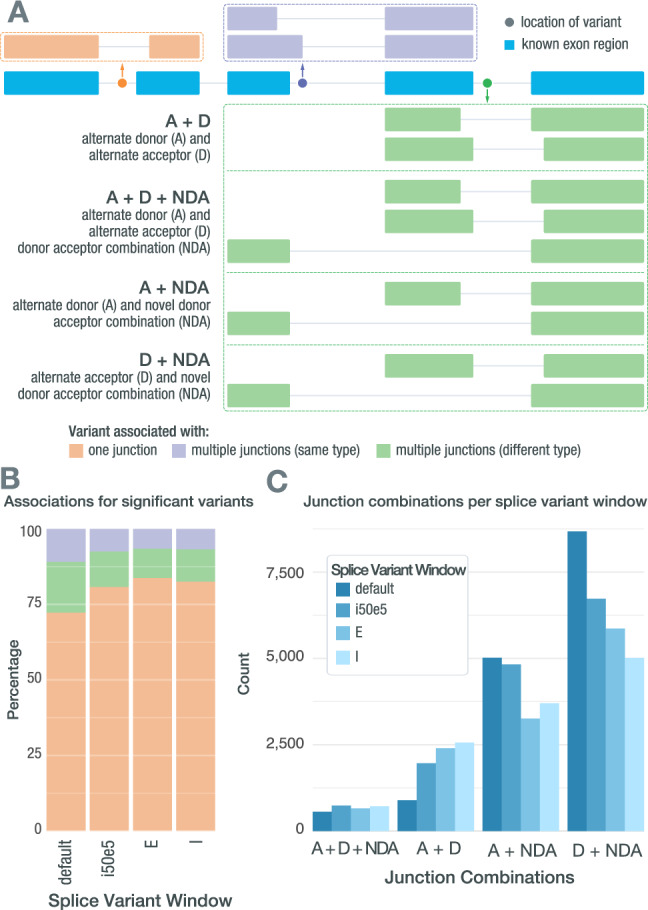
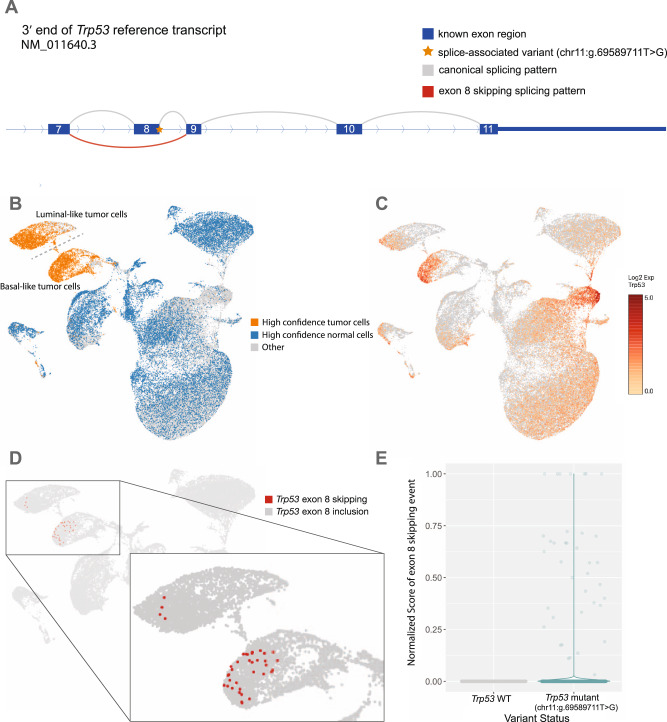
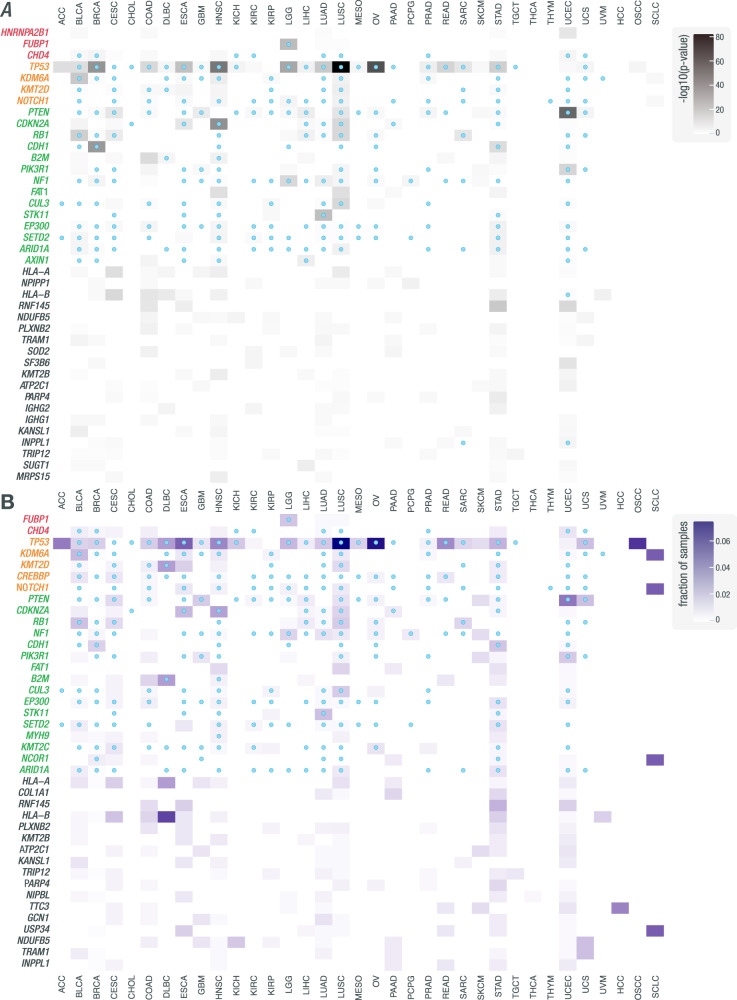
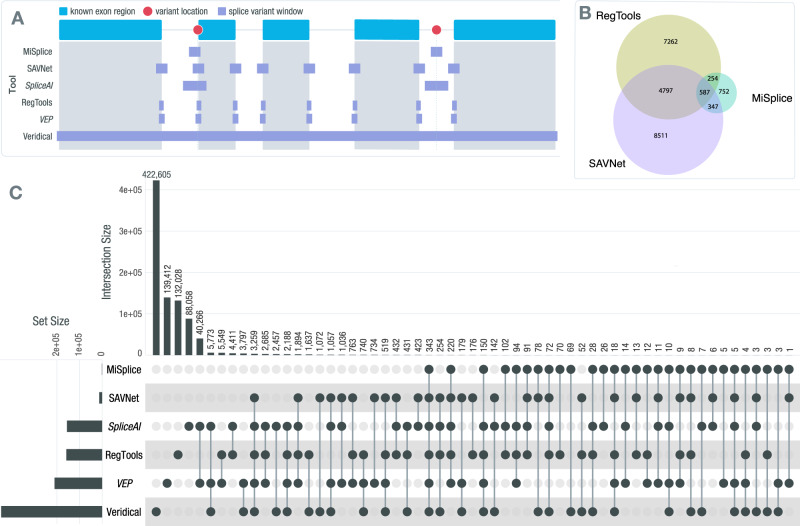
Somatic mutations within non-coding regions and even exons may have unidentified regulatory consequences that are often overlooked in analysis workflows. Here we present RegTools ( www.regtools.org ), a computationally efficient, free, and open-source software package designed to integrate somatic variants from genomic data with splice junctions from bulk or single cell transcriptomic data to identify variants that may cause aberrant splicing. We apply RegTools to over 9000 tumor samples with both tumor DNA and RNA sequence data. RegTools discovers 235,778 events where a splice-associated variant significantly increases the splicing of a particular junction, across 158,200 unique variants and 131,212 unique junctions. To characterize these somatic variants and their associated splice isoforms, we annotate them with the Variant Effect Predictor, SpliceAI, and Genotype-Tissue Expression junction counts and compare our results to other tools that integrate genomic and transcriptomic data. While many events are corroborated by the aforementioned tools, the flexibility of RegTools also allows us to identify splice-associated variants in known cancer drivers, such as TP53, CDKN2A, and B2M, and other genes.
© 2023. The Author(s).
Conflict of interest statement
W.C.C serves on the advisory board for Novartis Pharmaceutical and reports intellectual property with Pathfinder Therapeutics. R.U. reports grants and personal fees from Merck Inc. R.G. served as consultant for Horizon Pharmaceuticals and GenePlus. The remaining authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
