

Profiling of Circulating Tumor Cells for Screening of Selective Inhibitors of Tumor-Initiating Stem-Like Cells
- PMID: 36949364
- PMCID: PMC10190641
- DOI: 10.1002/advs.202206812
Profiling of Circulating Tumor Cells for Screening of Selective Inhibitors of Tumor-Initiating Stem-Like Cells
Retraction in
-
RETRACTION: Profiling of Circulating Tumor Cells for Screening of Selective Inhibitors of Tumor-Initiating Stem-Like Cells.Adv Sci (Weinh). 2025 May;12(18):e2505952. doi: 10.1002/advs.202505952. Epub 2025 Apr 11. Adv Sci (Weinh). 2025. PMID: 40211882 Free PMC article. No abstract available.
Abstract
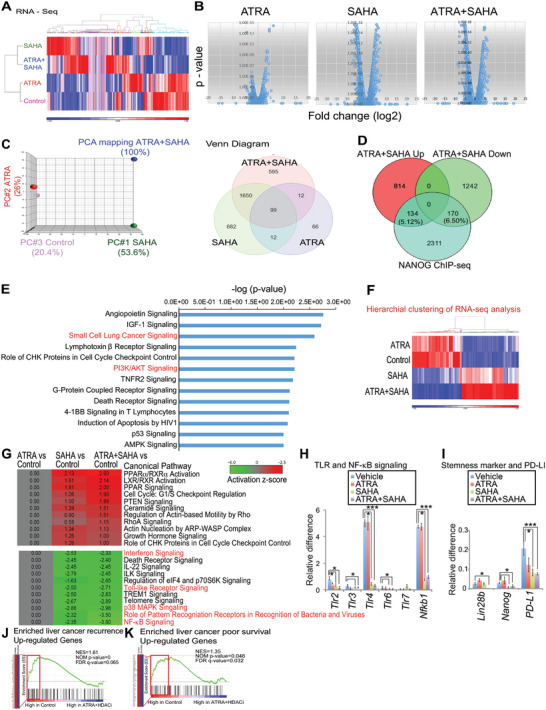
A critical barrier to effective cancer therapy is the improvement of drug selectivity, toxicity, and reduced recurrence of tumors expanded from tumor-initiating stem-like cells (TICs). The aim is to identify circulating tumor cell (CTC)-biomarkers and to identify an effective combination of TIC-specific, repurposed federal drug administration (FDA)-approved drugs. Three different types of high-throughput screens targeting the TIC population are employed: these include a CD133 (+) cell viability screen, a NANOG expression screen, and a drug combination screen. When combined in a refined secondary screening approach that targets Nanog expression with the same FDA-approved drug library, histone deacetylase (HDAC) inhibitor(s) combined with all-trans retinoic acid (ATRA) demonstrate the highest efficacy for inhibition of TIC growth in vitro and in vivo. Addition of immune checkpoint inhibitor further decreases recurrence and extends PDX mouse survival. RNA-seq analysis of TICs reveals that combined drug treatment reduces many Toll-like receptors (TLR) and stemness genes through repression of the lncRNA MIR22HG. This downregulation induces PTEN and TET2, leading to loss of the self-renewal property of TICs. Thus, CTC biomarker analysis would predict the prognosis and therapy response to this drug combination. In general, biomarker-guided stratification of HCC patients and TIC-targeted therapy should eradicate TICs to extend HCC patient survival.
Keywords: alcohol; hepatocellular carcinoma (HCC); tumor-initiating stem-like cells (TIC).
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Zhou B.‐B. S., Zhang H., Damelin M., Geles K. G., Grindley J. C., Dirks P. B., Nat. Rev. Drug Discovery 2009, 8, 806. - PubMed
-
- Suetsugu A., Nagaki M., Aoki H., Motohashi T., Kunisada T., Moriwaki H., Biochem. Biophys. Res. Commun. 2006, 351, 820. - PubMed
-
- Ma S., Lee T. K., Zheng B.‐J., Chan K. W., Guan X.‐Y., Oncogene 2008, 27, 1749. - PubMed
-
- Finn R. S., Qin S., Ikeda M., Galle P. R., Ducreux M., Kim T.‐Y., Kudo M., Breder V., Merle P., Kaseb A. O., Li D., Verret W., Xu D.‐Z., Hernandez S., Liu J., Huang C., Mulla S., Wang Y., Lim H. Y, Zhu A. X., Cheng A.‐L., N. Engl. J. Med. 2020, 382, 1894. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 1R01AA018857-01/GF/NIH HHS/United States
- R01 AA018857/AA/NIAAA NIH HHS/United States
- P50 AA011999/AA/NIAAA NIH HHS/United States
- R21 AA025470/AA/NIAAA NIH HHS/United States
- UL1 TR000130/TR/NCATS NIH HHS/United States
- 5P30DK048522-13/GF/NIH HHS/United States
- R21 AA025470-01A1/GF/NIH HHS/United States
- P50AA011999/GF/NIH HHS/United States
- R24 AA012885/AA/NIAAA NIH HHS/United States
- R01 AA025204-01A1/GF/NIH HHS/United States
- P30 CA014089/CA/NCI NIH HHS/United States
- R01 AA025204/AA/NIAAA NIH HHS/United States
- NH/NIH HHS/United States
- P30 DK048522/DK/NIDDK NIH HHS/United States
- UL1 TR001855/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials