

TP53- mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease
- PMID: 36949957
- PMCID: PMC10026701
- DOI: 10.1038/s44161-022-00206-6
TP53- mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease
Abstract
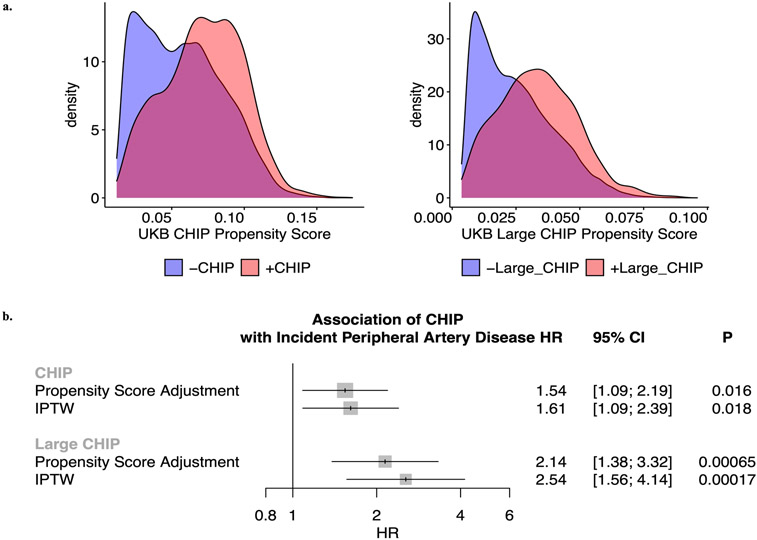
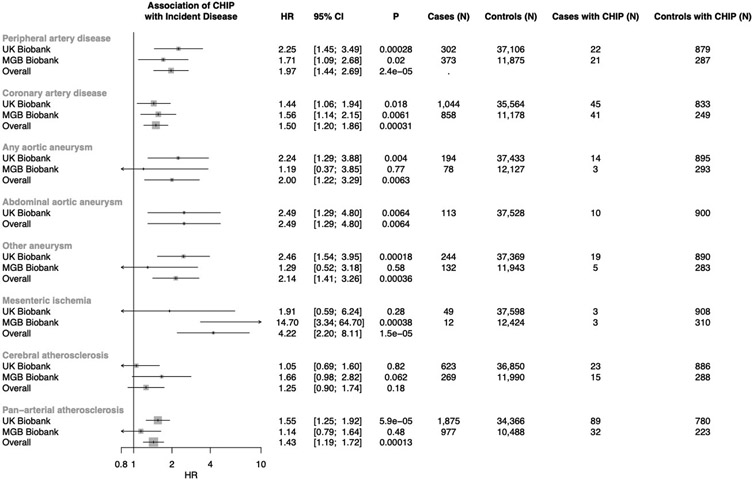
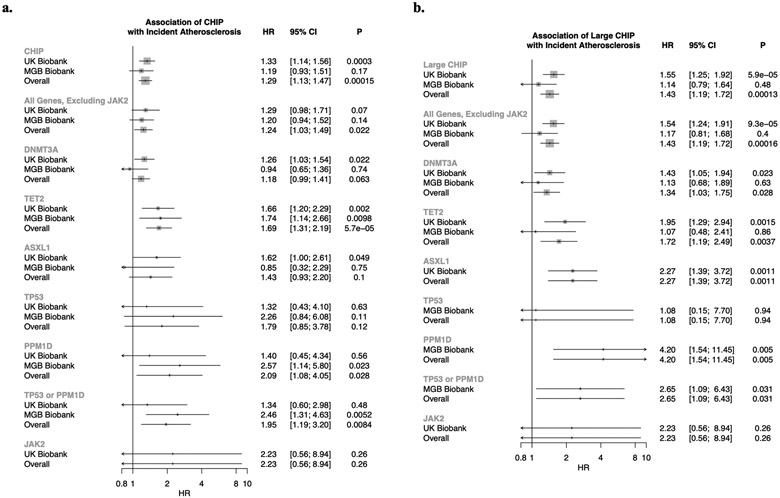
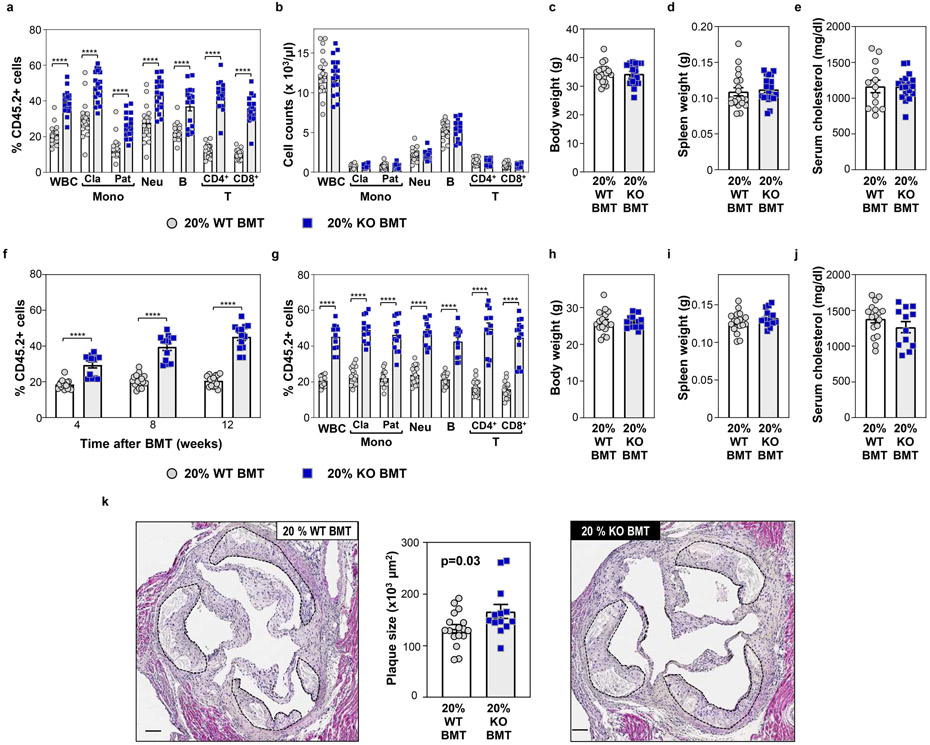
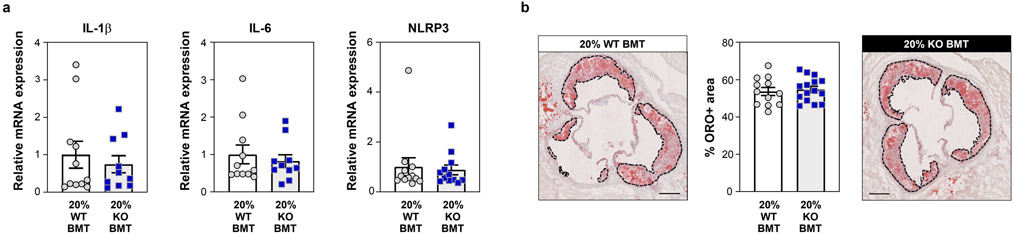
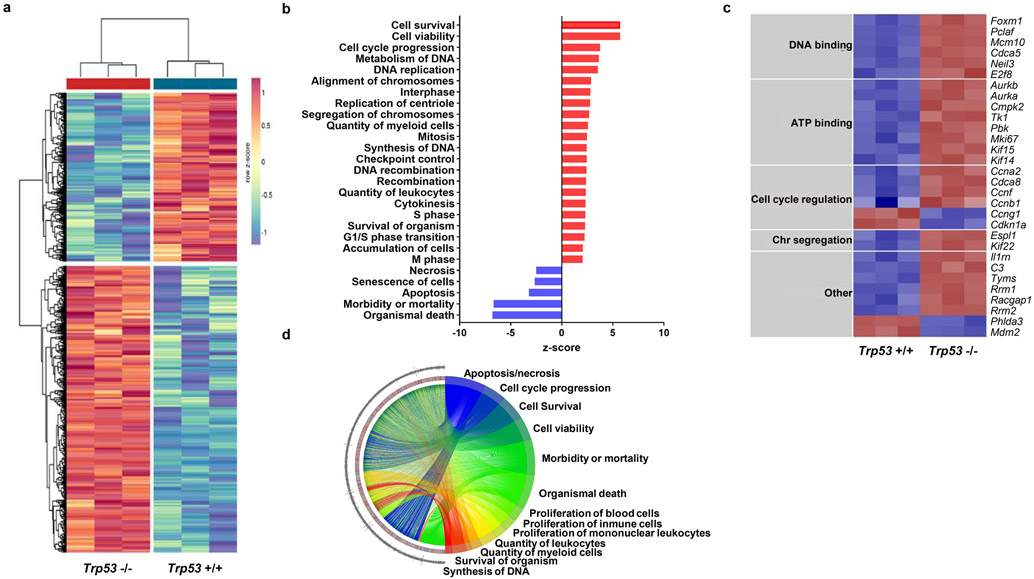
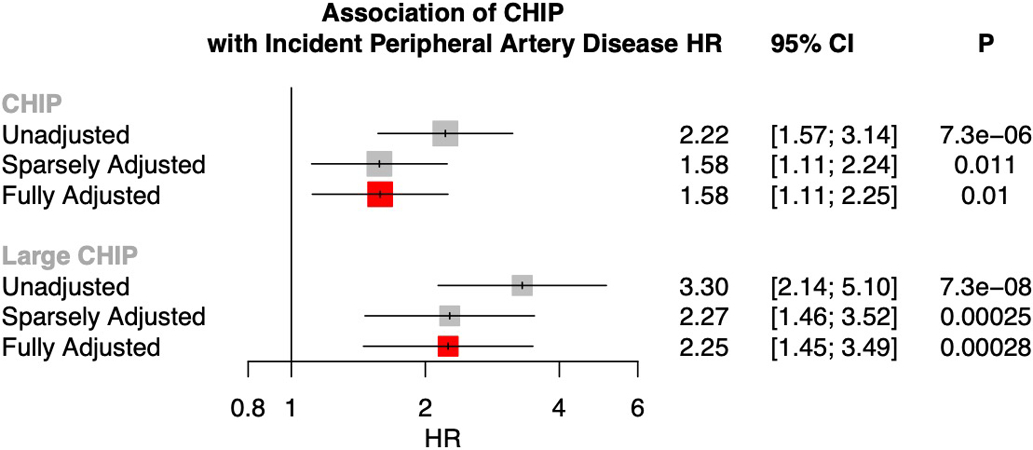
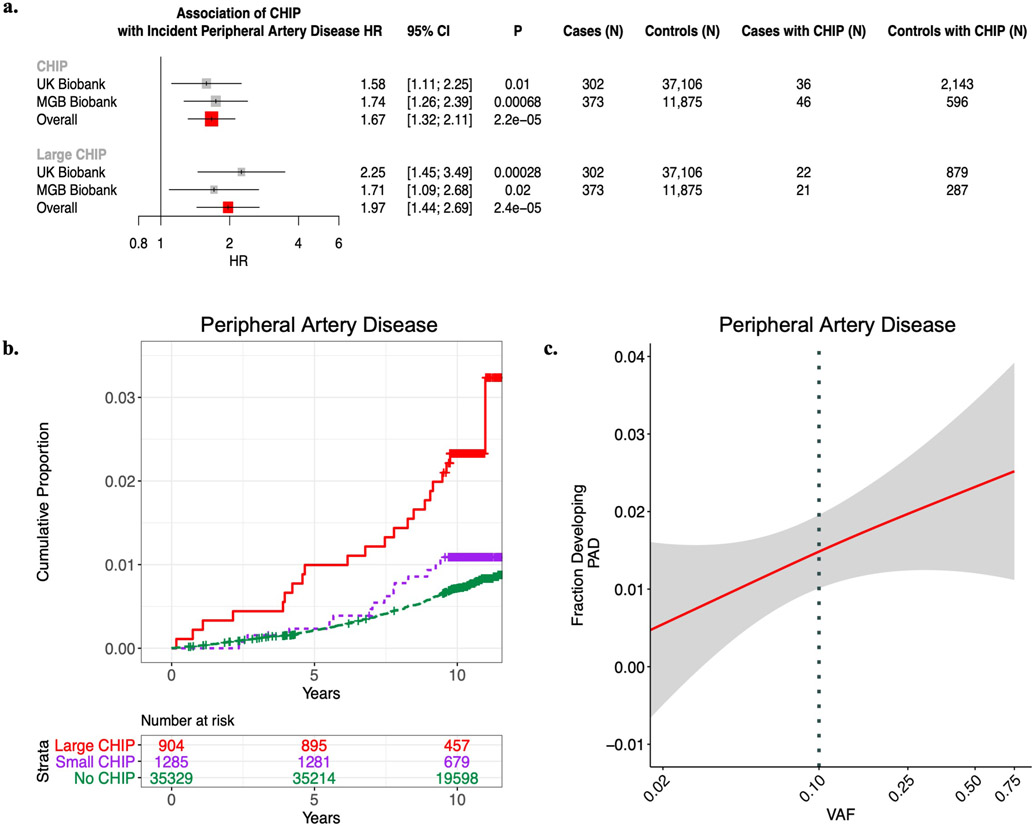
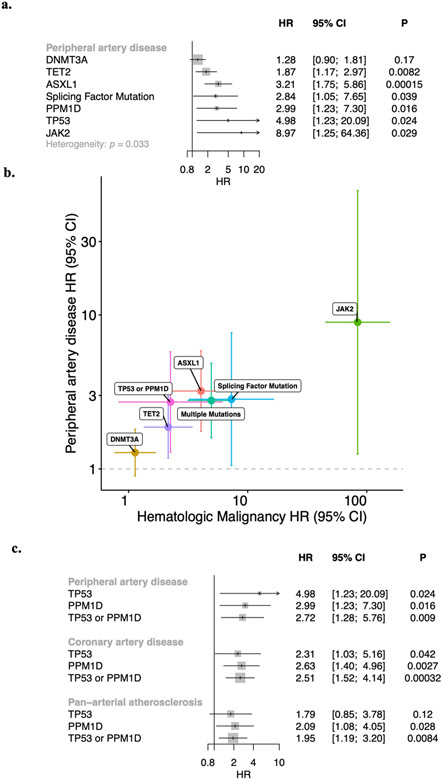
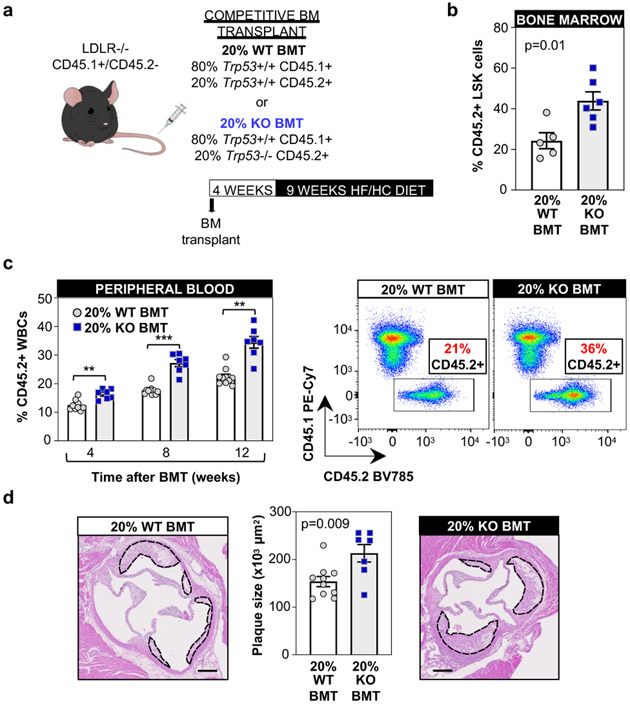
Somatic mutations in blood indicative of clonal hematopoiesis of indeterminate potential (CHIP) are associated with an increased risk of hematologic malignancy, coronary artery disease, and all-cause mortality. Here we analyze the relation between CHIP status and incident peripheral artery disease (PAD) and atherosclerosis, using whole-exome sequencing and clinical data from the UK Biobank and Mass General Brigham Biobank. CHIP associated with incident PAD and atherosclerotic disease across multiple beds, with increased risk among individuals with CHIP driven by mutation in DNA Damage Repair (DDR) genes such as TP53 and PPM1D. To model the effects of DDR-induced CHIP on atherosclerosis, we used a competitive bone marrow transplantation strategy, and generated atherosclerosis-prone Ldlr-/- chimeric mice carrying 20% p53-deficient hematopoietic cells. The chimeric mice were analyzed 13-weeks post-grafting and showed increased aortic plaque size and accumulation of macrophages within the plaque, driven by increased proliferation of p53-deficient plaque macrophages. In summary, our findings highlight the role of CHIP as a broad driver of atherosclerosis across the entire arterial system beyond the coronary arteries, and provide genetic and experimental support for a direct causal contribution of TP53-mutant CHIP to atherosclerosis.
Keywords: atherosclerosis; clonal hematopoiesis; sequencing; somatic.
Figures






Similar articles
-
Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease.N Engl J Med. 2017 Jul 13;377(2):111-121. doi: 10.1056/NEJMoa1701719. Epub 2017 Jun 21. N Engl J Med. 2017. PMID: 28636844 Free PMC article.
-
Clonal Hematopoiesis of Indeterminate Potential Predicts Adverse Outcomes in Patients With Atherosclerotic Cardiovascular Disease.J Am Coll Cardiol. 2023 May 23;81(20):1996-2009. doi: 10.1016/j.jacc.2023.03.401. J Am Coll Cardiol. 2023. PMID: 37197843 Free PMC article.
-
Leukocytes carrying Clonal Hematopoiesis of Indeterminate Potential (CHIP) Mutations invade Human Atherosclerotic Plaques.medRxiv [Preprint]. 2023 Jul 26:2023.07.22.23292754. doi: 10.1101/2023.07.22.23292754. medRxiv. 2023. PMID: 37546840 Free PMC article. Preprint.
-
Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease.J Mol Cell Cardiol. 2021 Dec;161:98-105. doi: 10.1016/j.yjmcc.2021.07.004. Epub 2021 Jul 21. J Mol Cell Cardiol. 2021. PMID: 34298011 Free PMC article. Review.
-
p53 involvement in clonal hematopoiesis of indeterminate potential.Curr Opin Hematol. 2019 Jul;26(4):235-240. doi: 10.1097/MOH.0000000000000509. Curr Opin Hematol. 2019. PMID: 31045645 Free PMC article. Review.
Cited by
-
Clonal hematopoiesis in cardiovascular disease and therapeutic implications.Nat Cardiovasc Res. 2022 Feb;1(2):116-124. doi: 10.1038/s44161-021-00015-3. Epub 2022 Feb 7. Nat Cardiovasc Res. 2022. PMID: 36337911 Free PMC article.
-
Somatic Mutations in Cardiovascular Disease.Circ Res. 2022 Jan 7;130(1):149-161. doi: 10.1161/CIRCRESAHA.121.319809. Epub 2022 Jan 7. Circ Res. 2022. PMID: 34995138 Free PMC article. Review.
-
Atherosclerotic burden and cerebral small vessel disease: exploring the link through microvascular aging and cerebral microhemorrhages.Geroscience. 2024 Oct;46(5):5103-5132. doi: 10.1007/s11357-024-01139-7. Epub 2024 Apr 19. Geroscience. 2024. PMID: 38639833 Free PMC article. Review.
-
Pathogenesis and inflammaging in myelodysplastic syndrome.Haematologica. 2025 Feb 1;110(2):283-299. doi: 10.3324/haematol.2023.284944. Haematologica. 2025. PMID: 39445405 Free PMC article. Review.
-
Association of clonal hematopoiesis of indeterminate potential with cardiometabolic multimorbidity progression and mortality: a prospective study of UK Biobank.Eur J Med Res. 2025 May 24;30(1):414. doi: 10.1186/s40001-025-02639-8. Eur J Med Res. 2025. PMID: 40410892 Free PMC article.
References
-
- Song P, et al. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: an updated systematic review and analysis. Lancet Glob Health 7, e1020–e1030 (2019). - PubMed
Grants and funding
- F30 HL149180/HL/NHLBI NIH HHS/United States
- DP5 OD029586/OD/NIH HHS/United States
- R01 HL163099/HL/NHLBI NIH HHS/United States
- T32 GM136651/GM/NIGMS NIH HHS/United States
- R01 HL148565/HL/NHLBI NIH HHS/United States
- R01 HL134892/HL/NHLBI NIH HHS/United States
- OT3 HL147154/HL/NHLBI NIH HHS/United States
- R01 HL080472/HL/NHLBI NIH HHS/United States
- T32 HL007208/HL/NHLBI NIH HHS/United States
- IK2 CX001780/CX/CSRD VA/United States
- IK2 BX005759/BX/BLRD VA/United States
- R01 HL148050/HL/NHLBI NIH HHS/United States
- K08 CA263555/CA/NCI NIH HHS/United States
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous