

The neuroprotective effects of targeting key factors of neuronal cell death in neurodegenerative diseases: The role of ER stress, oxidative stress, and neuroinflammation
- PMID: 36950516
- PMCID: PMC10025411
- DOI: 10.3389/fncel.2023.1105247
The neuroprotective effects of targeting key factors of neuronal cell death in neurodegenerative diseases: The role of ER stress, oxidative stress, and neuroinflammation
Abstract
Neuronal loss is one of the striking causes of various central nervous system (CNS) disorders, including major neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and Amyotrophic lateral sclerosis (ALS). Although these diseases have different features and clinical manifestations, they share some common mechanisms of disease pathology. Progressive regional loss of neurons in patients is responsible for motor, memory, and cognitive dysfunctions, leading to disabilities and death. Neuronal cell death in neurodegenerative diseases is linked to various pathways and conditions. Protein misfolding and aggregation, mitochondrial dysfunction, generation of reactive oxygen species (ROS), and activation of the innate immune response are the most critical hallmarks of most common neurodegenerative diseases. Thus, endoplasmic reticulum (ER) stress, oxidative stress, and neuroinflammation are the major pathological factors of neuronal cell death. Even though the exact mechanisms are not fully discovered, the notable role of mentioned factors in neuronal loss is well known. On this basis, researchers have been prompted to investigate the neuroprotective effects of targeting underlying pathways to determine a promising therapeutic approach to disease treatment. This review provides an overview of the role of ER stress, oxidative stress, and neuroinflammation in neuronal cell death, mainly discussing the neuroprotective effects of targeting pathways or molecules involved in these pathological factors.
Keywords: ER stress; ROS; UPR – unfolded protein response; cell death; neurodegenerative diseases; neuroinflammation; oxidative stress.
Copyright © 2023 Karvandi, Sheikhzadeh Hesari, Aref and Mahdavi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
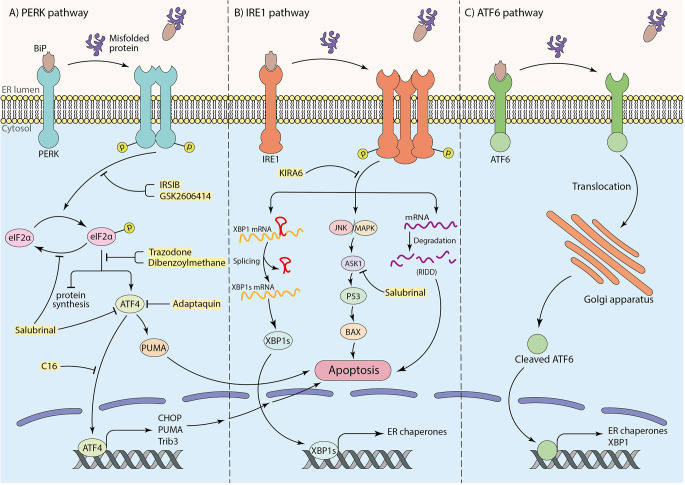
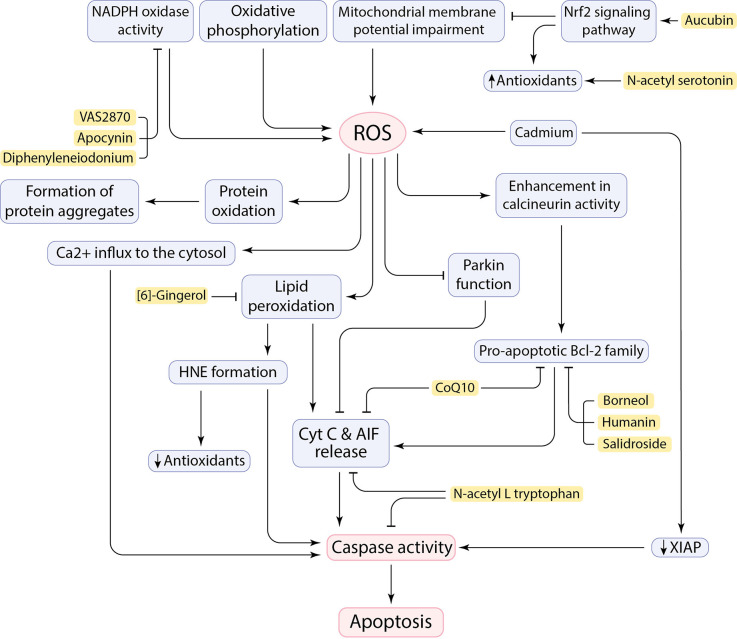
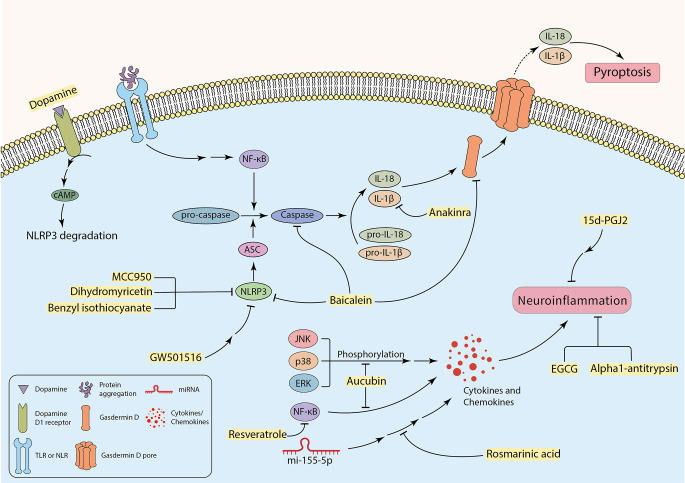
References
-
- Aime P., Karuppagounder S. S., Rao A., Chen Y., Burke R. E., Ratan R. R., et al. . (2020). The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson’s disease. Neurobiol. Dis. 136:104725. 10.1016/j.nbd.2019.104725 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous