

Blockade of Activin Receptor IIB Protects Arthritis Pathogenesis by Non-Amplification of Activin A-ACVR2B-NOX4 Axis Pathway
- PMID: 36950748
- PMCID: PMC10190289
- DOI: 10.1002/advs.202205161
Blockade of Activin Receptor IIB Protects Arthritis Pathogenesis by Non-Amplification of Activin A-ACVR2B-NOX4 Axis Pathway
Abstract
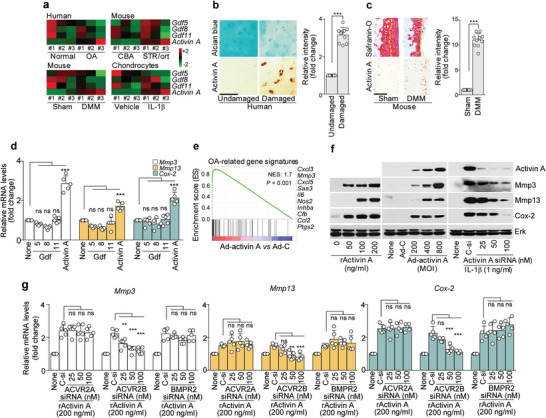
Although activin receptor IIB (ACVR2B) is emerging as a novel pathogenic receptor, its ligand and assembled components (or assembly) are totally unknown in the context of osteoarthritis (OA) pathogenesis. The present results suggest that upregulation of ACVR2B and its assembly could affect osteoarthritic cartilage destruction. It is shown that the ACVR2B ligand, activin A, regulates catabolic factor expression through ACVR2B in OA development. Activin A Tg mice (Col2a1-Inhba) exhibit enhanced cartilage destruction, whereas heterozygous activin A KO mice (Inhba+/- ) show protection from cartilage destruction. In silico analysis suggests that the Activin A-ACVR2B axis is involved in Nox4-dependent ROS production. Activin A Tg:Nox4 KO (Col2a1-Inhba:Nox4-/- ) mice show inhibition of experimental OA pathogenesis. NOX4 directly binds to the C-terminal binding site on ACVR2B-ACVR1B and amplifies the pathogenic signal for cartilage destruction through SMAD2/3 signaling. Together, the findings reveal that the ACVR2B assembly, which comprises Activin A, ACVR2B, ACVR1B, Nox4, and AP-1-induced HIF-2α, accelerates OA development. Furthermore, it is shown that shRNA-mediated ACVR2B knockdown or trapping ligands of ACVR2B abrogate OA development by competitively disrupting the ACVR2B-Activin A interaction. These results suggest that the ACVR2B assembly is required to amplify osteoarthritic cartilage destruction and could be a potential therapeutic target in efforts to treat OA.
Keywords: ACVR2B assembly; arthritis treatment; druggable target; human OA cartilage; mouse model.
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Hunter D. J., Schofield D., Callander E., Nat. Rev. Rheumatol. 2014, 17, 437. - PubMed
-
- Heinegard D., Saxne T., Nat. Rev. Rheumatol. 2011, 7, 50. - PubMed
-
- Blom A. B., van Lent P. L., Libregts S., Holthuysen A. E., van der Kraan P. M., van Rooijen N., van den Berg W. B., Arthritis Rheum. 2007, 56, 147. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- CRC21021/the National Research Council of Science & Technology (NST) of the Korean government (MSIT)
- SRC-2017R1A5A1014560/National Research Foundation of Korea
- NRF-2021M3E5E7023855/National Research Foundation of Korea
- NRF-2022R1A2C1004688/National Research Foundation of Korea
- NRF-2022R1A2C2004343/National Research Foundation of Korea
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous