

On the Origin and Evolution of Microbial Mercury Methylation
- PMID: 36951100
- PMCID: PMC10083202
- DOI: 10.1093/gbe/evad051
On the Origin and Evolution of Microbial Mercury Methylation
Abstract
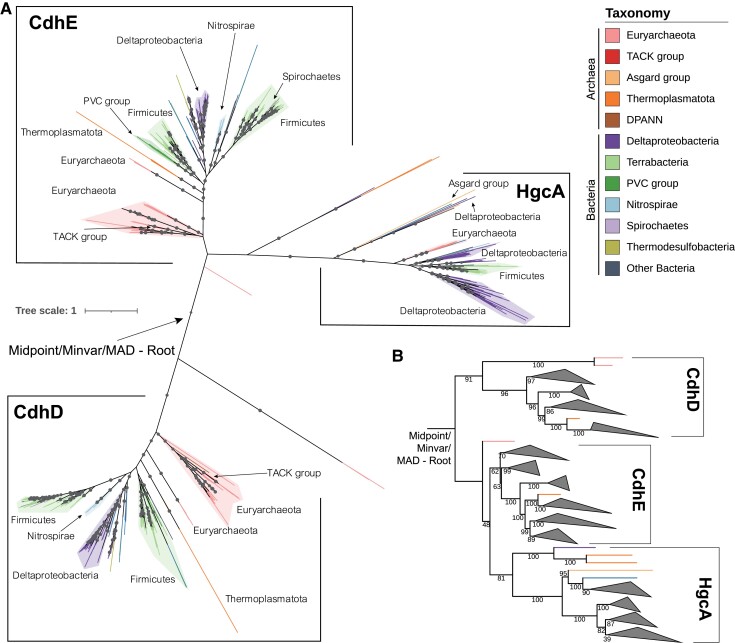
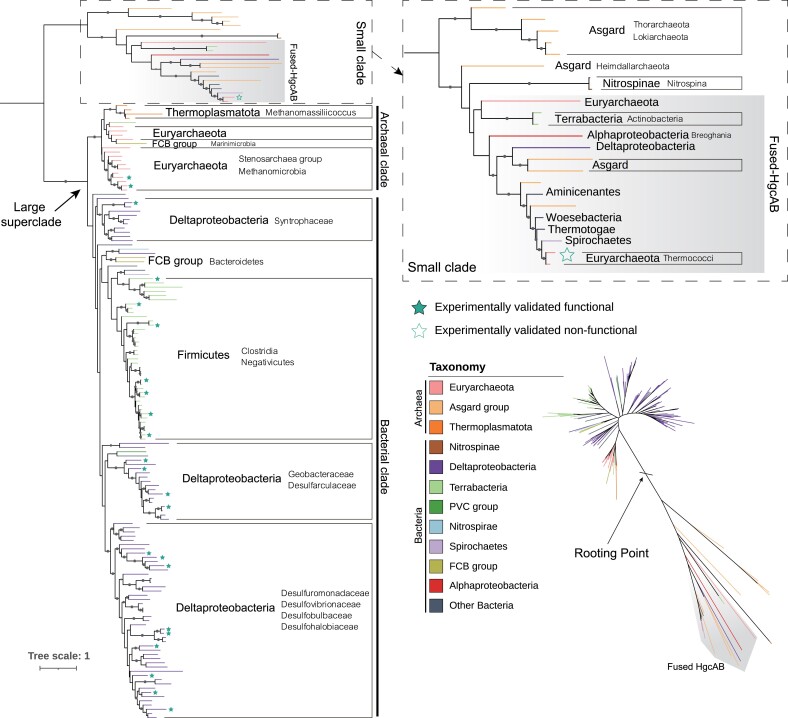
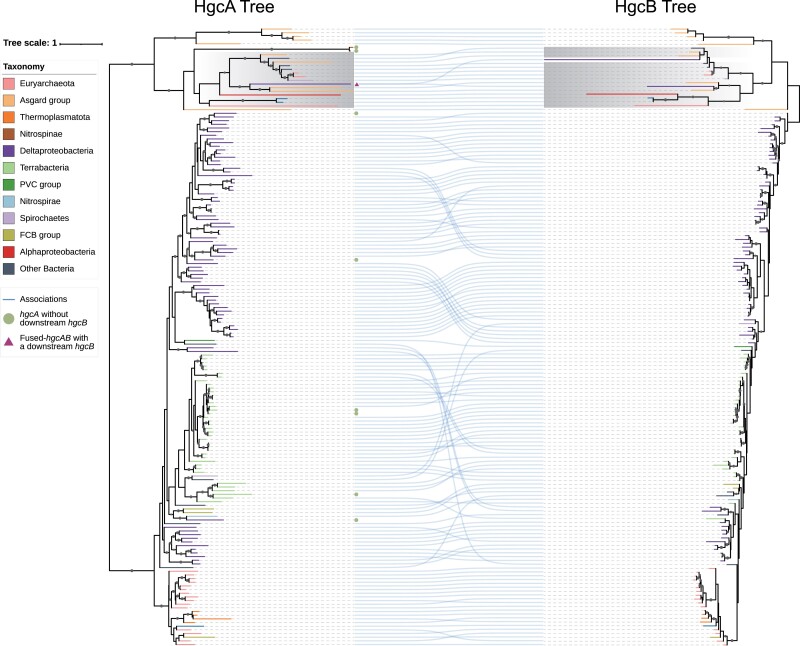
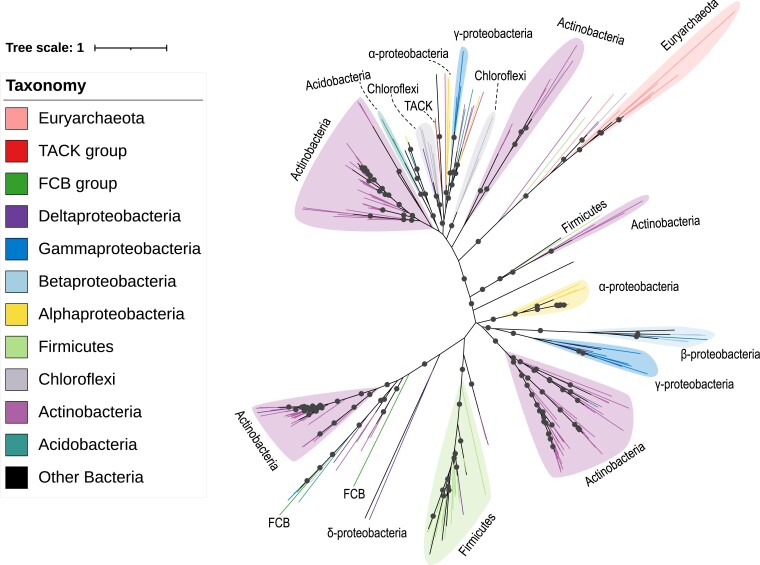
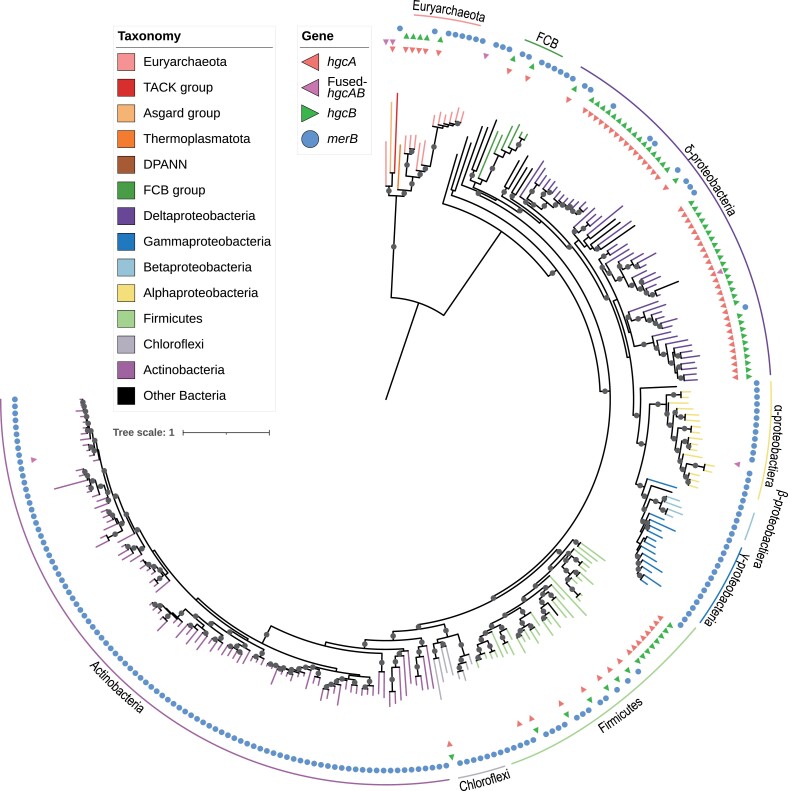
The origin of microbial mercury methylation has long been a mystery. Here, we employed genome-resolved phylogenetic analyses to decipher the evolution of the mercury-methylating gene, hgcAB, constrain the ancestral origin of the hgc operon, and explain the distribution of hgc in Bacteria and Archaea. We infer the extent to which vertical inheritance and horizontal gene transfer have influenced the evolution of mercury methylators and hypothesize that evolution of this trait bestowed the ability to produce an antimicrobial compound (MeHg+) on a potentially resource-limited early Earth. We speculate that, in response, the evolution of MeHg+-detoxifying alkylmercury lyase (encoded by merB) reduced a selective advantage for mercury methylators and resulted in widespread loss of hgc in Bacteria and Archaea.
Keywords: hgc gene; LUCA; antimicrobial; evolution; gene loss; horizontal gene transfer; mercury; methylmercury.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
Similar articles
-
Robust Mercury Methylation across Diverse Methanogenic Archaea.mBio. 2018 Apr 10;9(2):e02403-17. doi: 10.1128/mBio.02403-17. mBio. 2018. PMID: 29636434 Free PMC article.
-
Carbon Amendments Alter Microbial Community Structure and Net Mercury Methylation Potential in Sediments.Appl Environ Microbiol. 2018 Jan 17;84(3):e01049-17. doi: 10.1128/AEM.01049-17. Print 2018 Feb 1. Appl Environ Microbiol. 2018. PMID: 29150503 Free PMC article.
-
Periphyton and Flocculent Materials Are Important Ecological Compartments Supporting Abundant and Diverse Mercury Methylator Assemblages in the Florida Everglades.Appl Environ Microbiol. 2019 Jun 17;85(13):e00156-19. doi: 10.1128/AEM.00156-19. Print 2019 Jul 1. Appl Environ Microbiol. 2019. PMID: 31028023 Free PMC article.
-
Microbial mercury transformations: Molecules, functions and organisms.Adv Appl Microbiol. 2022;118:31-90. doi: 10.1016/bs.aambs.2022.03.001. Epub 2022 Apr 13. Adv Appl Microbiol. 2022. PMID: 35461663 Review.
-
Microbial Mercury Methylation in Aquatic Environments: A Critical Review of Published Field and Laboratory Studies.Environ Sci Technol. 2019 Jan 2;53(1):4-19. doi: 10.1021/acs.est.8b02709. Epub 2018 Dec 21. Environ Sci Technol. 2019. PMID: 30525497 Review.
Cited by
-
The role of prokaryotic mercury methylators and demethylators in Canadian Arctic thermokarst lakes.Sci Rep. 2025 Feb 28;15(1):7173. doi: 10.1038/s41598-025-89438-7. Sci Rep. 2025. PMID: 40021694 Free PMC article.
-
Mining-impacted rice paddies select for Archaeal methylators and reveal a putative (Archaeal) regulator of mercury methylation.ISME Commun. 2023 Jul 15;3(1):74. doi: 10.1038/s43705-023-00277-x. ISME Commun. 2023. PMID: 37454192 Free PMC article.
-
Functional Genes and Transcripts Indicate the Existent and Active Microbial Mercury-Methylating Community in Mangrove Intertidal Sediments of an Urbanized Bay.Microorganisms. 2024 Jun 20;12(6):1245. doi: 10.3390/microorganisms12061245. Microorganisms. 2024. PMID: 38930626 Free PMC article.
References
-
- Amyot M, Morel FM, Ariya PA. 2005. Dark oxidation of dissolved and liquid elemental mercury in aquatic environments. Environ Sci Technol. 39(1):110–114. - PubMed
-
- Barkay T, Kritee K, Boyd E, Geesey G. 2010. A thermophilic bacterial origin and subsequent constraints by redox, light and salinity on the evolution of the microbial mercuric reductase. Environ Microbiol. 12:2904–2917. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
