

Global population structure, genomic diversity and carbohydrate fermentation characteristics of clonal complex 119 (CC119), an understudied Shiga toxin-producing E. coli (STEC) lineage including O165:H25 and O172:H25
- PMID: 36951916
- PMCID: PMC10132054
- DOI: 10.1099/mgen.0.000959
Global population structure, genomic diversity and carbohydrate fermentation characteristics of clonal complex 119 (CC119), an understudied Shiga toxin-producing E. coli (STEC) lineage including O165:H25 and O172:H25
Abstract
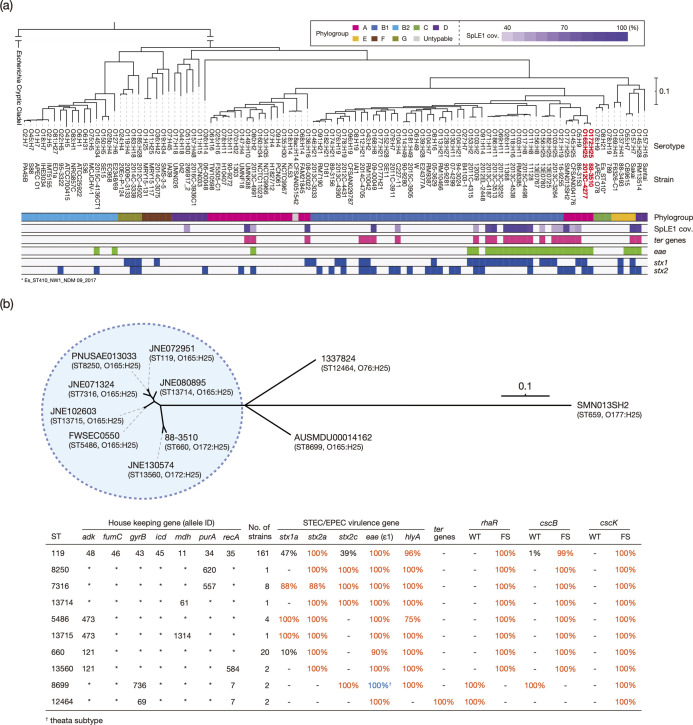
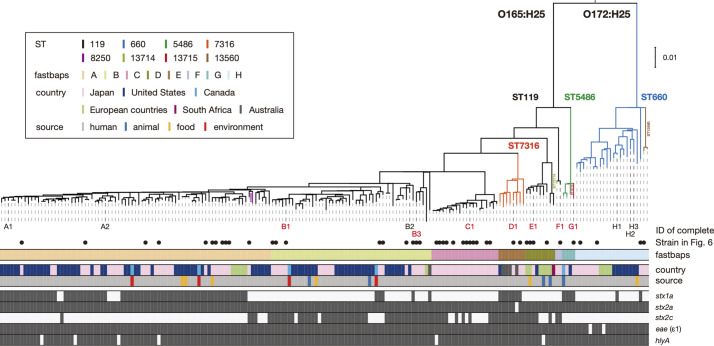
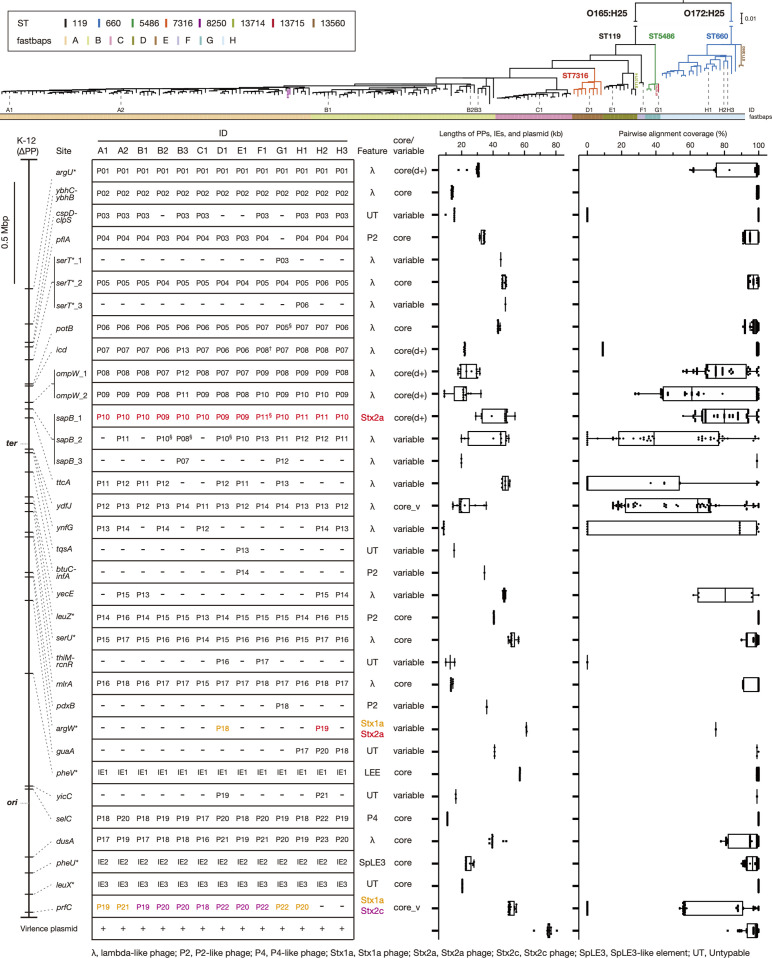
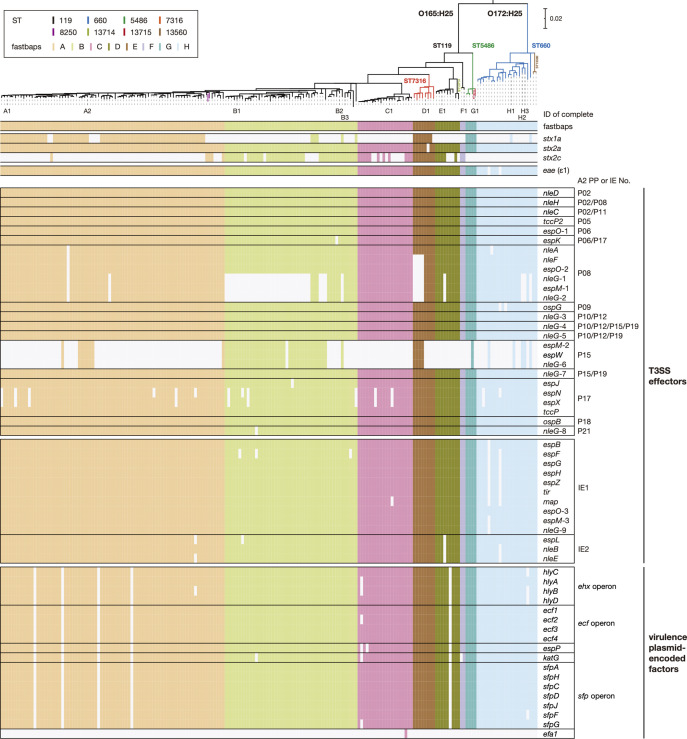
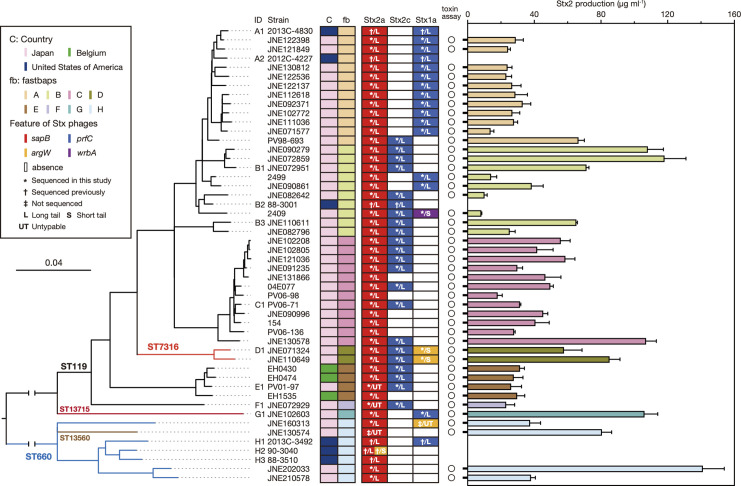
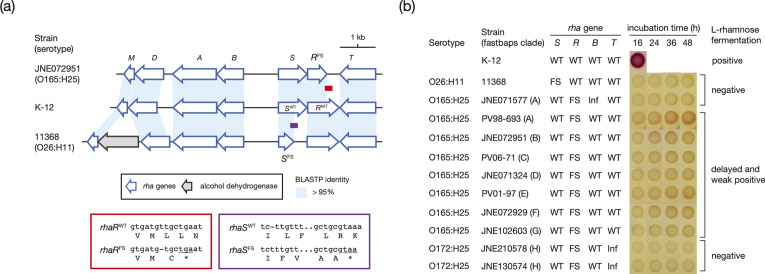
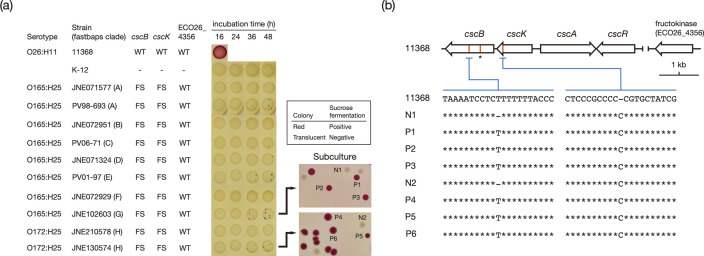
Among Shiga toxin (Stx)-producing Escherichia coli (STEC) strains of various serotypes, O157:H7 and five major non-O157 STEC (O26:H11, O111:H8, O103:H2, O121:H19 and O145:H28) can be selectively isolated by using tellurite-containing media. While human infections by O165:H25 STEC strains have been reported worldwide, their detection and isolation are not easy, as they are not resistant to tellurite. Systematic whole-genome sequencing (WGS) analyses have not yet been conducted. Here, we defined O165:H25 strains and their close relatives, including O172:H25 strains, as clonal complex 119 (CC119) and performed a global WGS analysis of the major lineage of CC119, called CC119 sensu stricto (CC119ss), by using 202 CC119ss strains, including 90 strains sequenced in this study. Detailed comparisons of 13 closed genomes, including 7 obtained in this study, and systematic analyses of Stx phage genomes in 50 strains covering the entire CC119ss lineage, were also conducted. These analyses revealed that the Stx2a phage, the locus of enterocyte effacement (LEE) encoding a type III secretion system (T3SS), many prophages encoding T3SS effectors, and the virulence plasmid were acquired by the common ancestor of CC119ss and have been stably maintained in this lineage, while unusual exchanges of Stx1a and Stx2c phages were found at a single integration site. Although the genome sequences of Stx2a phages were highly conserved, CC119ss strains exhibited notable variation in Stx2 production levels. Further analyses revealed the lack of SpLE1-like elements carrying the tellurite resistance genes in CC119ss and defects in rhamnose, sucrose, salicin and dulcitol fermentation. The genetic backgrounds underlying these defects were also clarified.
Keywords: O165:H25; O172:H25; Shiga toxin-producing Escherichia coli; carbohydrate fermentation; comparative genomics; phylogenetic analysis.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures

Similar articles
-
The global population structure and evolutionary history of the acquisition of major virulence factor-encoding genetic elements in Shiga toxin-producing Escherichia coli O121:H19.Microb Genom. 2021 Dec;7(12):000716. doi: 10.1099/mgen.0.000716. Microb Genom. 2021. PMID: 34878971 Free PMC article.
-
Detection of Shiga toxin-producing Escherichia coli serotypes O26:H11, O103:H2, O111:H8, O145:H28, and O157:H7 in raw-milk cheeses by using multiplex real-time PCR.Appl Environ Microbiol. 2011 Mar;77(6):2035-41. doi: 10.1128/AEM.02089-10. Epub 2011 Jan 14. Appl Environ Microbiol. 2011. PMID: 21239543 Free PMC article.
-
Comparative genomic analysis of a Shiga toxin-producing Escherichia coli (STEC) O145:H25 associated with a severe pediatric case of hemolytic uremic syndrome in Davidson County, Tennessee, US.BMC Genomics. 2020 Aug 17;21(1):564. doi: 10.1186/s12864-020-06967-3. BMC Genomics. 2020. PMID: 32807093 Free PMC article.
-
Low-density macroarray targeting non-locus of enterocyte effacement effectors (nle genes) and major virulence factors of Shiga toxin-producing Escherichia coli (STEC): a new approach for molecular risk assessment of STEC isolates.Appl Environ Microbiol. 2010 Jan;76(1):203-11. doi: 10.1128/AEM.01921-09. Epub 2009 Oct 30. Appl Environ Microbiol. 2010. PMID: 19880649 Free PMC article.
-
Evolution of virulence factors in Shiga-toxin-producing Escherichia coli.Cell Mol Life Sci. 1999 Nov 30;56(9-10):735-41. doi: 10.1007/s000180050020. Cell Mol Life Sci. 1999. PMID: 11212333 Free PMC article. Review.
Cited by
-
Exposure of Helicobacter pylori to clarithromycin in vitro resulting in the development of resistance and triggers metabolic reprogramming associated with virulence and pathogenicity.PLoS One. 2024 Mar 6;19(3):e0298434. doi: 10.1371/journal.pone.0298434. eCollection 2024. PLoS One. 2024. PMID: 38446753 Free PMC article.
-
Diversity of Shiga toxin transducing phages in Escherichia coli O145:H28 and the different Shiga toxin 2 production levels associated with short- or long-tailed phages.Front Microbiol. 2024 Aug 6;15:1453887. doi: 10.3389/fmicb.2024.1453887. eCollection 2024. Front Microbiol. 2024. PMID: 39165568 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources