

Global detection of human variants and isoforms by deep proteome sequencing
- PMID: 36959352
- PMCID: PMC10713452
- DOI: 10.1038/s41587-023-01714-x
Global detection of human variants and isoforms by deep proteome sequencing
Abstract
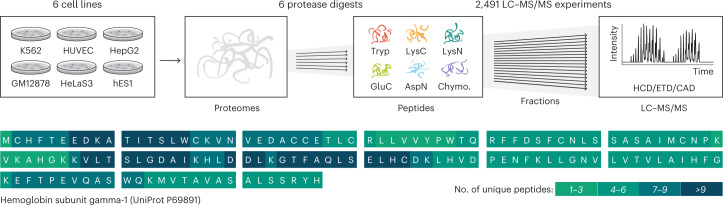
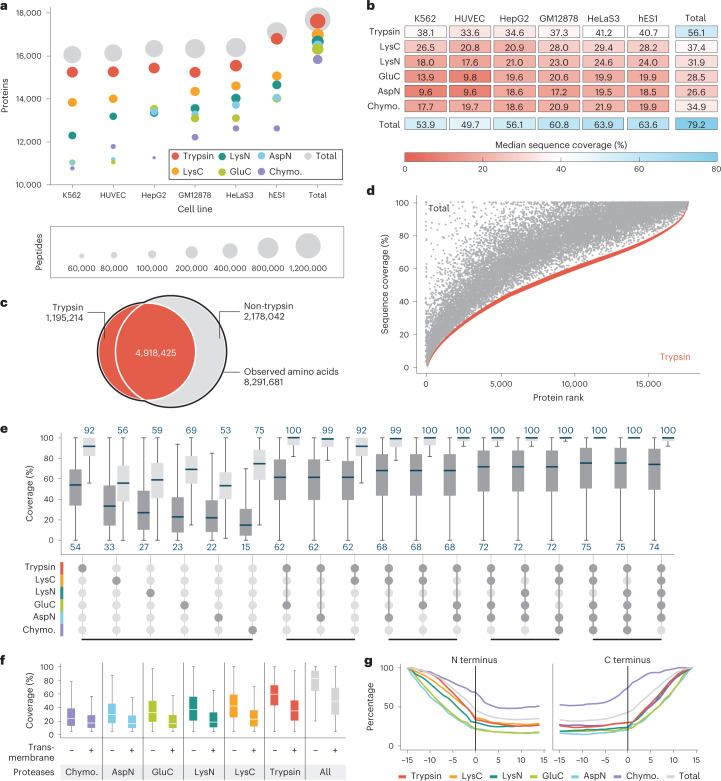
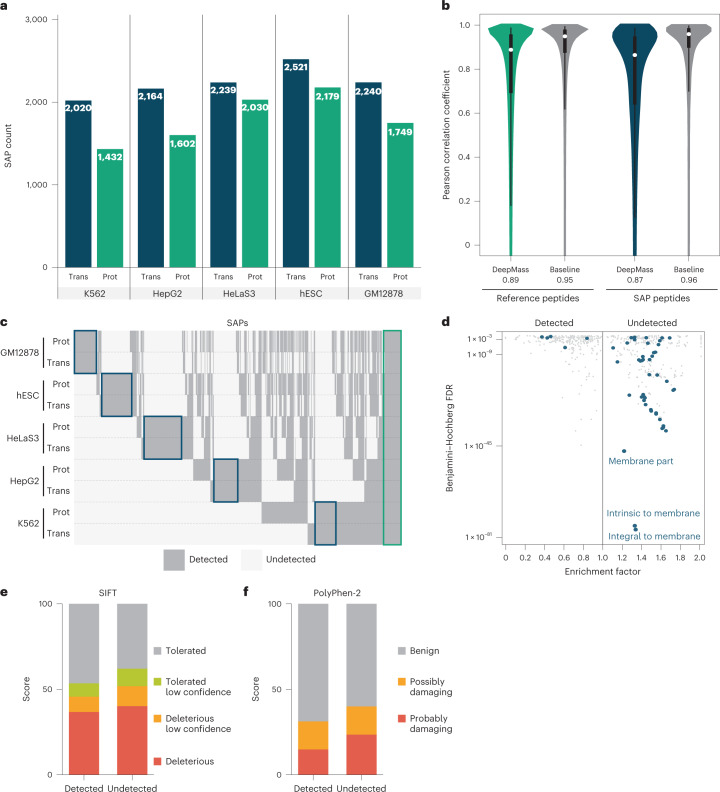
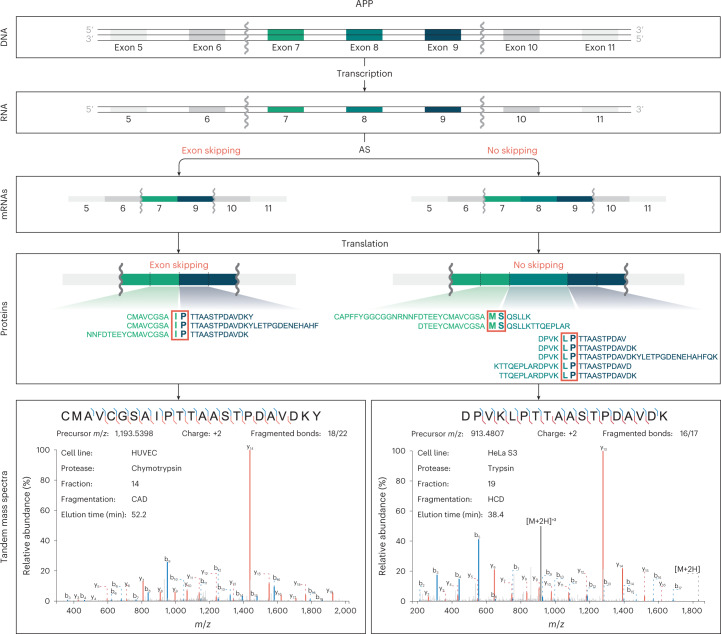
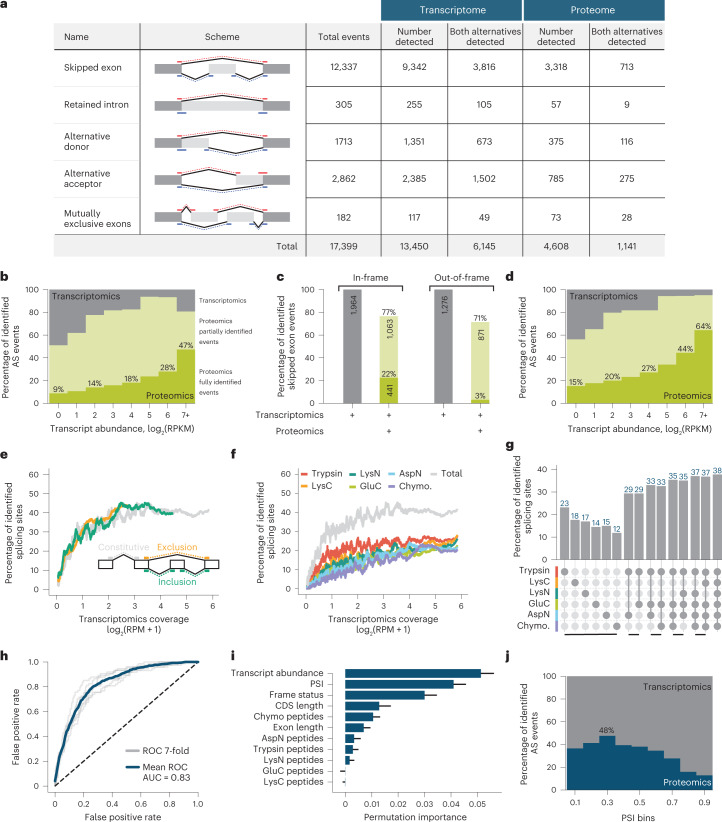
An average shotgun proteomics experiment detects approximately 10,000 human proteins from a single sample. However, individual proteins are typically identified by peptide sequences representing a small fraction of their total amino acids. Hence, an average shotgun experiment fails to distinguish different protein variants and isoforms. Deeper proteome sequencing is therefore required for the global discovery of protein isoforms. Using six different human cell lines, six proteases, deep fractionation and three tandem mass spectrometry fragmentation methods, we identify a million unique peptides from 17,717 protein groups, with a median sequence coverage of approximately 80%. Direct comparison with RNA expression data provides evidence for the translation of most nonsynonymous variants. We have also hypothesized that undetected variants likely arise from mutation-induced protein instability. We further observe comparable detection rates for exon-exon junction peptides representing constitutive and alternative splicing events. Our dataset represents a resource for proteoform discovery and provides direct evidence that most frame-preserving alternatively spliced isoforms are translated.
© 2023. The Author(s).
Conflict of interest statement
J.J.C. is a consultant for Thermo Fisher Scientific, 908 Devices, and Seer. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
