

Regulated cell death pathways in kidney disease
- PMID: 36959481
- PMCID: PMC10035496
- DOI: 10.1038/s41581-023-00694-0
Regulated cell death pathways in kidney disease
Abstract
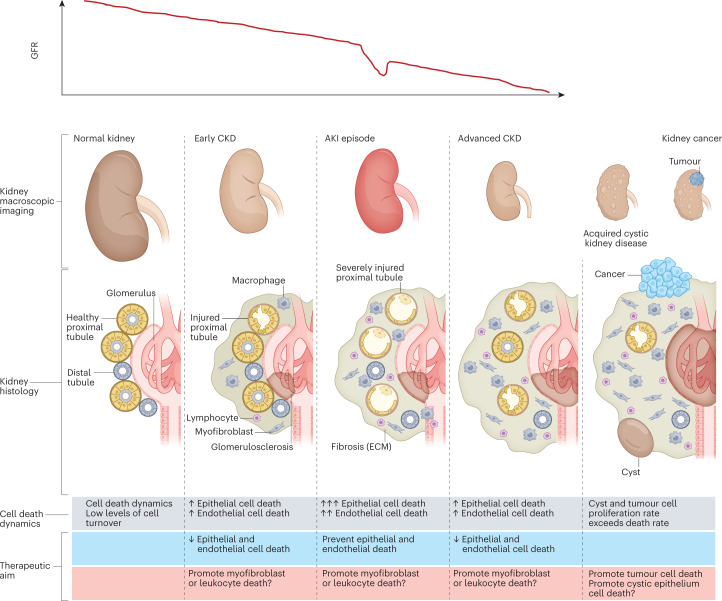
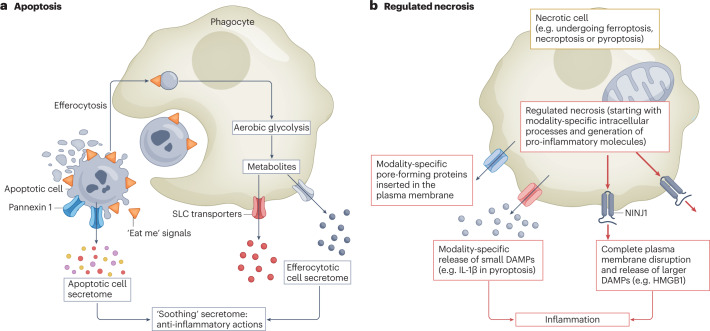
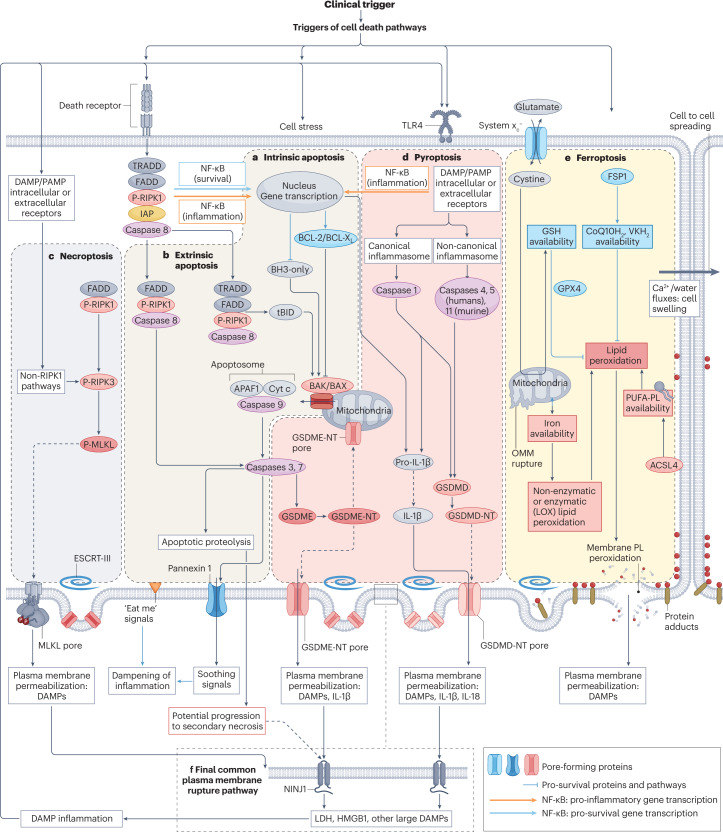
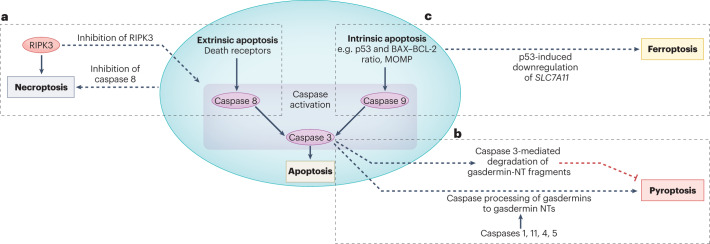
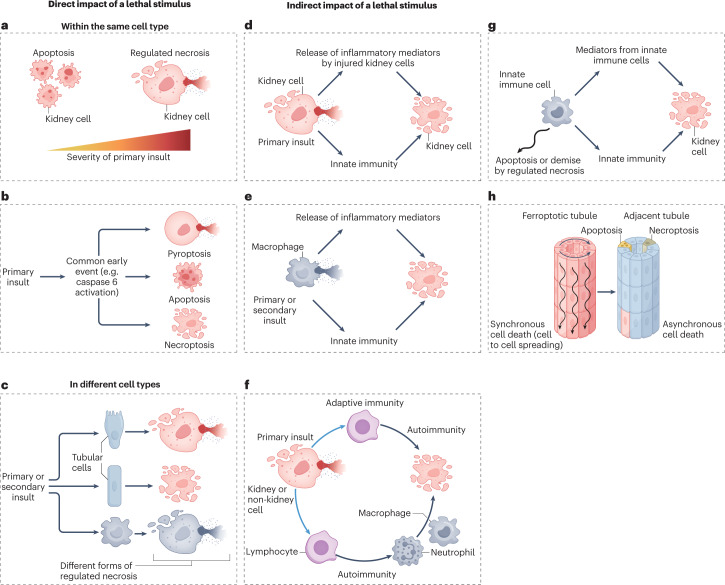
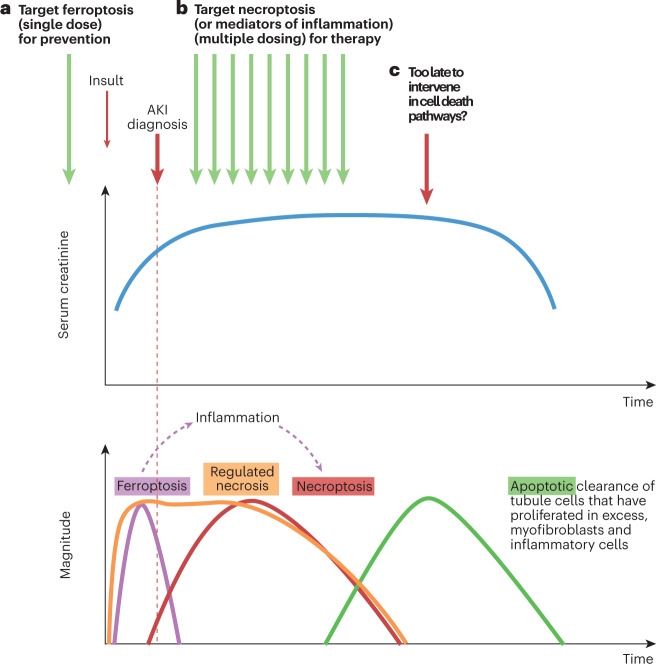
Disorders of cell number that result from an imbalance between the death of parenchymal cells and the proliferation or recruitment of maladaptive cells contributes to the pathogenesis of kidney disease. Acute kidney injury can result from an acute loss of kidney epithelial cells. In chronic kidney disease, loss of kidney epithelial cells leads to glomerulosclerosis and tubular atrophy, whereas interstitial inflammation and fibrosis result from an excess of leukocytes and myofibroblasts. Other conditions, such as acquired cystic disease and kidney cancer, are characterized by excess numbers of cyst wall and malignant cells, respectively. Cell death modalities act to clear unwanted cells, but disproportionate responses can contribute to the detrimental loss of kidney cells. Indeed, pathways of regulated cell death - including apoptosis and necrosis - have emerged as central events in the pathogenesis of various kidney diseases that may be amenable to therapeutic intervention. Modes of regulated necrosis, such as ferroptosis, necroptosis and pyroptosis may cause kidney injury directly or through the recruitment of immune cells and stimulation of inflammatory responses. Importantly, multiple layers of interconnections exist between different modalities of regulated cell death, including shared triggers, molecular components and protective mechanisms.
© 2023. Springer Nature Limited.
Conflict of interest statement
A.O. has received grants from Sanofi and consultancy, speaker fees or travel support from Advicciene, Astellas, AstraZeneca, Amicus, Amgen, Fresenius Medical Care, GSK, Bayer, Sanofi-Genzyme, Menarini, Kyowa Kirin, Alexion, Idorsia, Chiesi, Otsuka, Novo-Nordisk and Vifor Fresenius Medical Care Renal Pharma, and is the Director of the Catedra Mundipharma-UAM for diabetic kidney disease and the Catedra Astrazeneca-UAM for chronic kidney disease and electrolytes. A.O. also has stock in Telara Farma. The other authors declare no competing interests.
Figures
References
-
- Foreman KJ, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet. 2018;392:2052–2090. doi: 10.1016/S0140-6736(18)31694-5. - DOI - PMC - PubMed
-
- Chapman, A. B., Rahbari-Oskoui, F. F & Bennett, W. M. Acquired cystic disease of the kidney in adults. UpToDatehttps://www.uptodate.com/contents/acquired-cystic-disease-of-the-kidney-... (2023).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
