

Pediatric Nemaline Myopathy: A Systematic Review Using Individual Patient Data
- PMID: 36960434
- PMCID: PMC10032635
- DOI: 10.1177/08830738221096316
Pediatric Nemaline Myopathy: A Systematic Review Using Individual Patient Data
Abstract
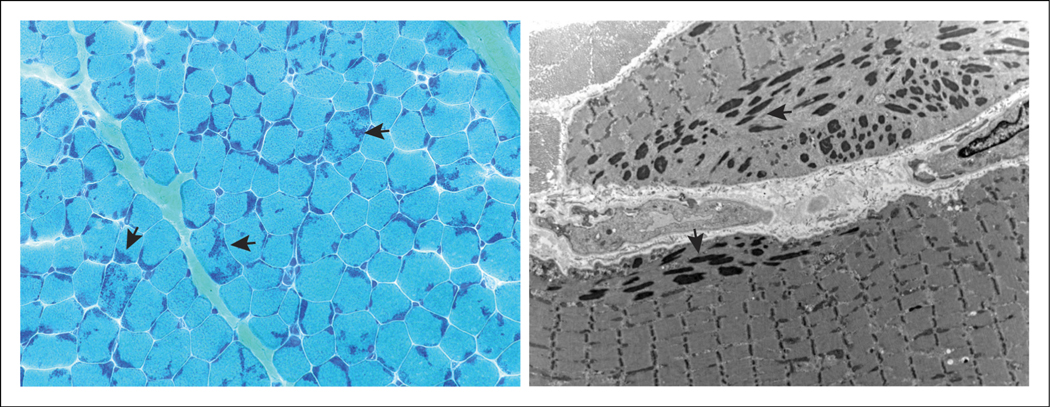
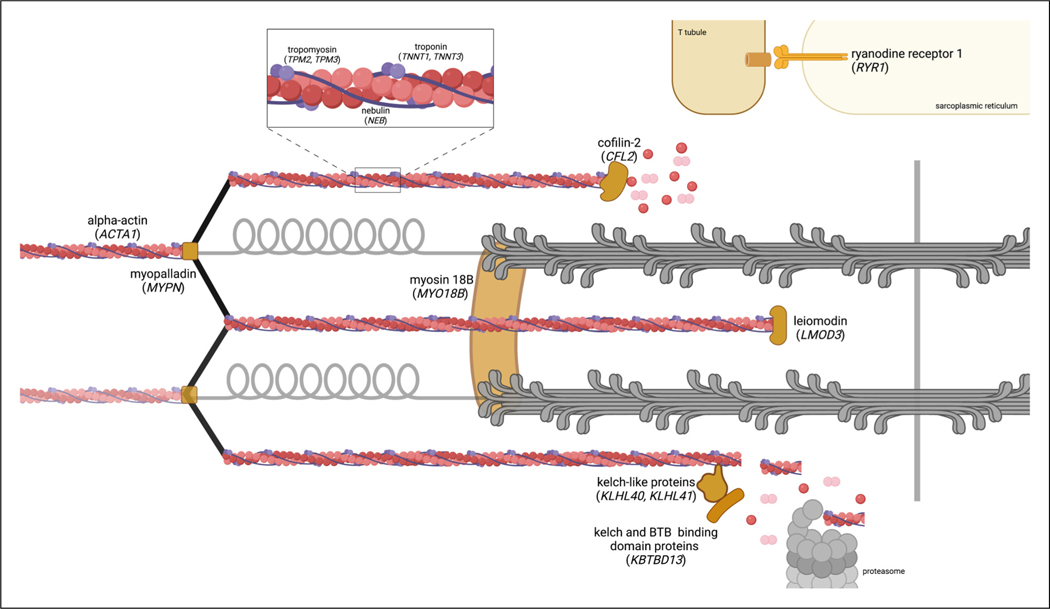
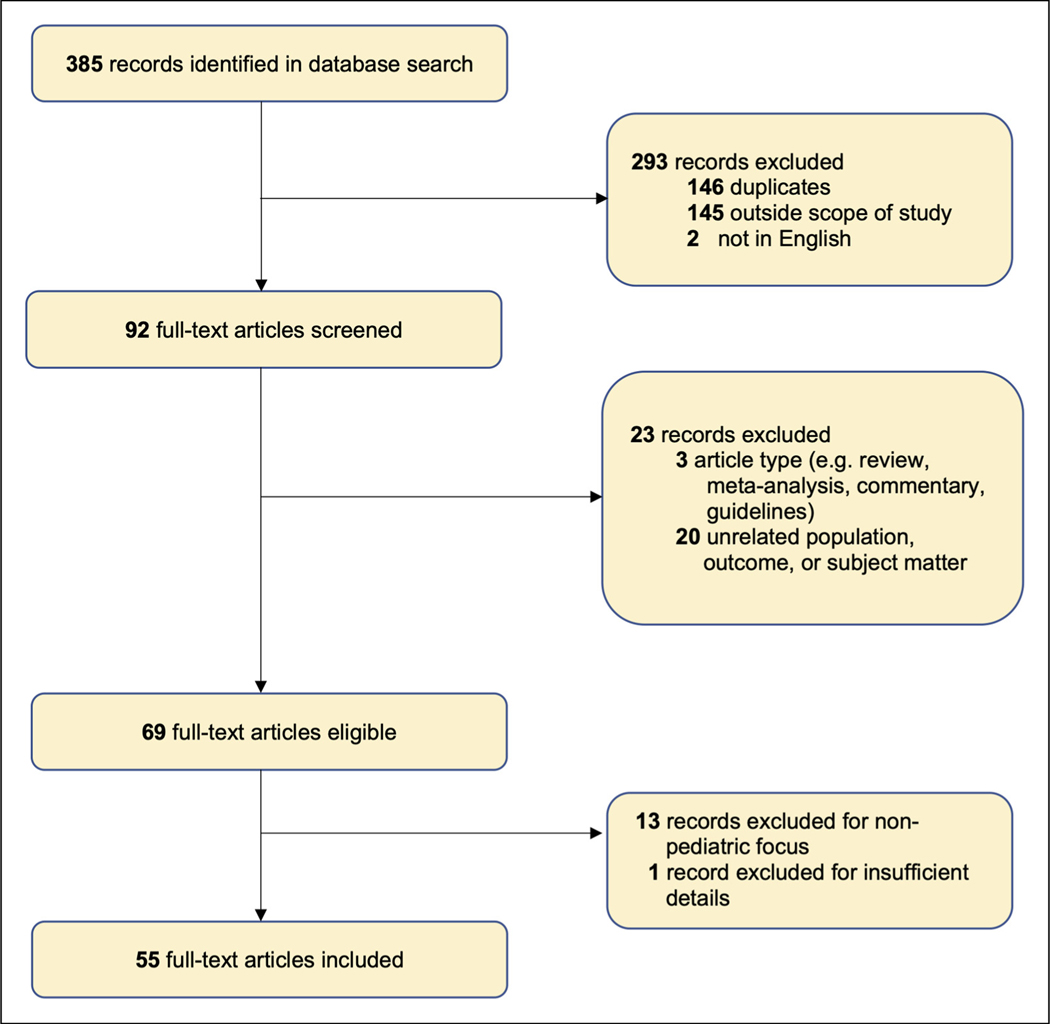
Nemaline myopathy is a skeletal muscle disease that affects 1 in 50 000 live births. The objective of this study was to develop a narrative synthesis of the findings of a systematic review of the latest case descriptions of patients with NM. A systematic search of MEDLINE, Embase, CINAHL, Web of Science, and Scopus was performed using Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guidelines using the keywords pediatric, child, NM, nemaline rod, and rod myopathy. Case studies focused on pediatric NM and published in English between January 1, 2010, and December 31, 2020, in order to represent the most recent findings. Information was collected about the age of first signs, earliest presenting neuromuscular signs and symptoms, systems affected, progression, death, pathologic description, and genetic changes. Of a total of 385 records, 55 case reports or series were reviewed, covering 101 pediatric patients from 23 countries. We review varying presentations in children ranging in severity despite being caused by the same mutation, in addition to current and future clinical considerations relevant to the care of patients with NM. This review synthesizes genetic, histopathologic, and disease presentation findings from pediatric NM case reports. These data strengthen our understanding of the wide spectrum of disease seen in NM. Future studies are needed to identify the underlying molecular mechanism of pathology, to improve diagnostics, and to develop better methods to improve the quality of life for these patients.
Keywords: muscle; myopathy; nemaline myopathy; sarcomere.
Conflict of interest statement
Declaration of Conflicting Interests The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures
References
-
- Wallgren-Pettersson C. Congenital Nemaline Myopathy: A Longitudinal Study. Finnish Society of Sciences and Letters; 1990.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
