

The Immunopeptidome from a Genomic Perspective: Establishing the Noncanonical Landscape of MHC Class I-Associated Peptides
- PMID: 36961404
- PMCID: PMC10236148
- DOI: 10.1158/2326-6066.CIR-22-0621
The Immunopeptidome from a Genomic Perspective: Establishing the Noncanonical Landscape of MHC Class I-Associated Peptides
Abstract
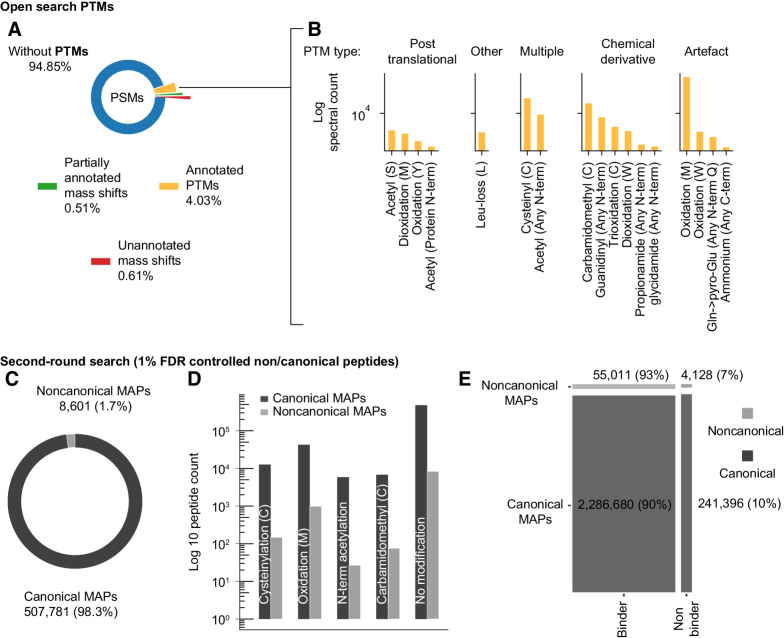
Tumor antigens can emerge through multiple mechanisms, including translation of noncoding genomic regions. This noncanonical category of tumor antigens has recently gained attention; however, our understanding of how they recur within and between cancer types is still in its infancy. Therefore, we developed a proteogenomic pipeline based on deep learning de novo mass spectrometry (MS) to enable the discovery of noncanonical MHC class I-associated peptides (ncMAP) from noncoding regions. Considering that the emergence of tumor antigens can also involve posttranslational modifications (PTM), we included an open search component in our pipeline. Leveraging the wealth of MS-based immunopeptidomics, we analyzed data from 26 MHC class I immunopeptidomic studies across 11 different cancer types. We validated the de novo identified ncMAPs, along with the most abundant PTMs, using spectral matching and controlled their FDR to 1%. The noncanonical presentation appeared to be 5 times enriched for the A03 HLA supertype, with a projected population coverage of 55%. The data reveal an atlas of 8,601 ncMAPs with varying levels of cancer selectivity and suggest 17 cancer-selective ncMAPs as attractive therapeutic targets according to a stringent cutoff. In summary, the combination of the open-source pipeline and the atlas of ncMAPs reported herein could facilitate the identification and screening of ncMAPs as targets for T-cell therapies or vaccine development.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
