

Mechanisms of obesity- and diabetes mellitus-related pancreatic carcinogenesis: a comprehensive and systematic review
- PMID: 36964133
- PMCID: PMC10039087
- DOI: 10.1038/s41392-023-01376-w
Mechanisms of obesity- and diabetes mellitus-related pancreatic carcinogenesis: a comprehensive and systematic review
Abstract
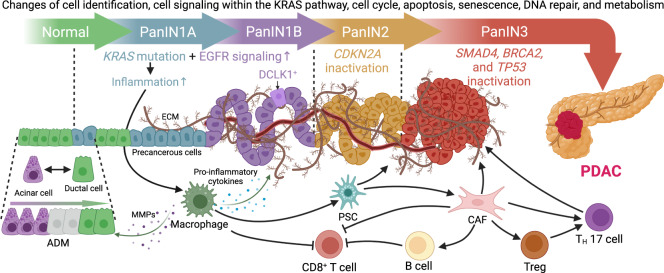
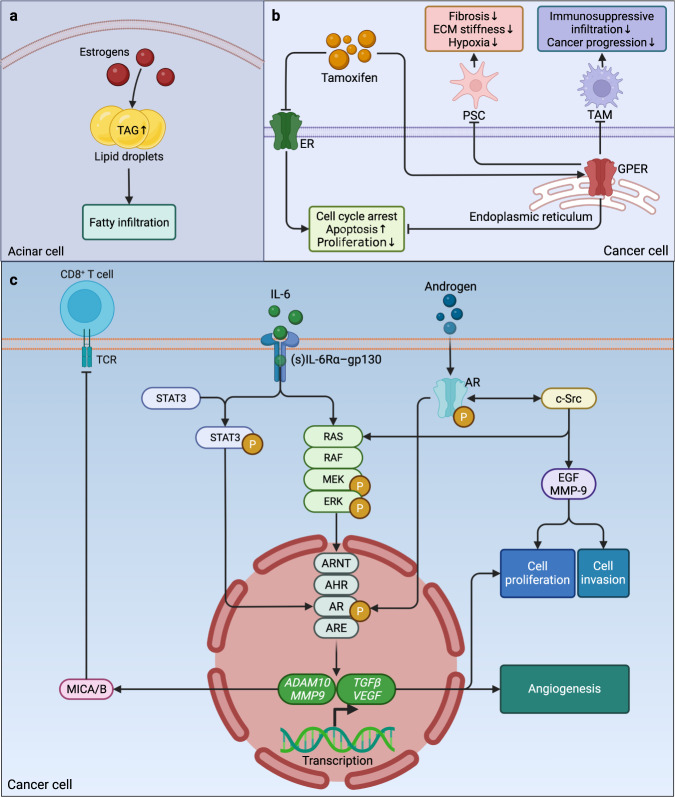
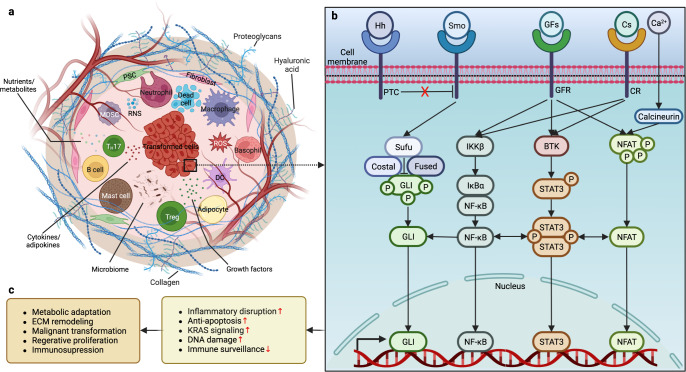
Research on obesity- and diabetes mellitus (DM)-related carcinogenesis has expanded exponentially since these two diseases were recognized as important risk factors for cancers. The growing interest in this area is prominently actuated by the increasing obesity and DM prevalence, which is partially responsible for the slight but constant increase in pancreatic cancer (PC) occurrence. PC is a highly lethal malignancy characterized by its insidious symptoms, delayed diagnosis, and devastating prognosis. The intricate process of obesity and DM promoting pancreatic carcinogenesis involves their local impact on the pancreas and concurrent whole-body systemic changes that are suitable for cancer initiation. The main mechanisms involved in this process include the excessive accumulation of various nutrients and metabolites promoting carcinogenesis directly while also aggravating mutagenic and carcinogenic metabolic disorders by affecting multiple pathways. Detrimental alterations in gastrointestinal and sex hormone levels and microbiome dysfunction further compromise immunometabolic regulation and contribute to the establishment of an immunosuppressive tumor microenvironment (TME) for carcinogenesis, which can be exacerbated by several crucial pathophysiological processes and TME components, such as autophagy, endoplasmic reticulum stress, oxidative stress, epithelial-mesenchymal transition, and exosome secretion. This review provides a comprehensive and critical analysis of the immunometabolic mechanisms of obesity- and DM-related pancreatic carcinogenesis and dissects how metabolic disorders impair anticancer immunity and influence pathophysiological processes to favor cancer initiation.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






References
-
- World Obesity Federation. World Obesity Atlas 2022 (World Obesity Federation, 2022).
-
- International Diabetes Federation. IDF Diabetes Atlas (International Diabetes Federation, Brussels, Belgium, 2021).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
