

A ciprofloxacin derivative with four mechanisms of action overcomes paclitaxel resistance in p53-mutant and MDR1 gene-expressing type II human endometrial cancer
- PMID: 36965280
- PMCID: PMC10092294
- DOI: 10.1016/j.biomaterials.2023.122093
A ciprofloxacin derivative with four mechanisms of action overcomes paclitaxel resistance in p53-mutant and MDR1 gene-expressing type II human endometrial cancer
Abstract
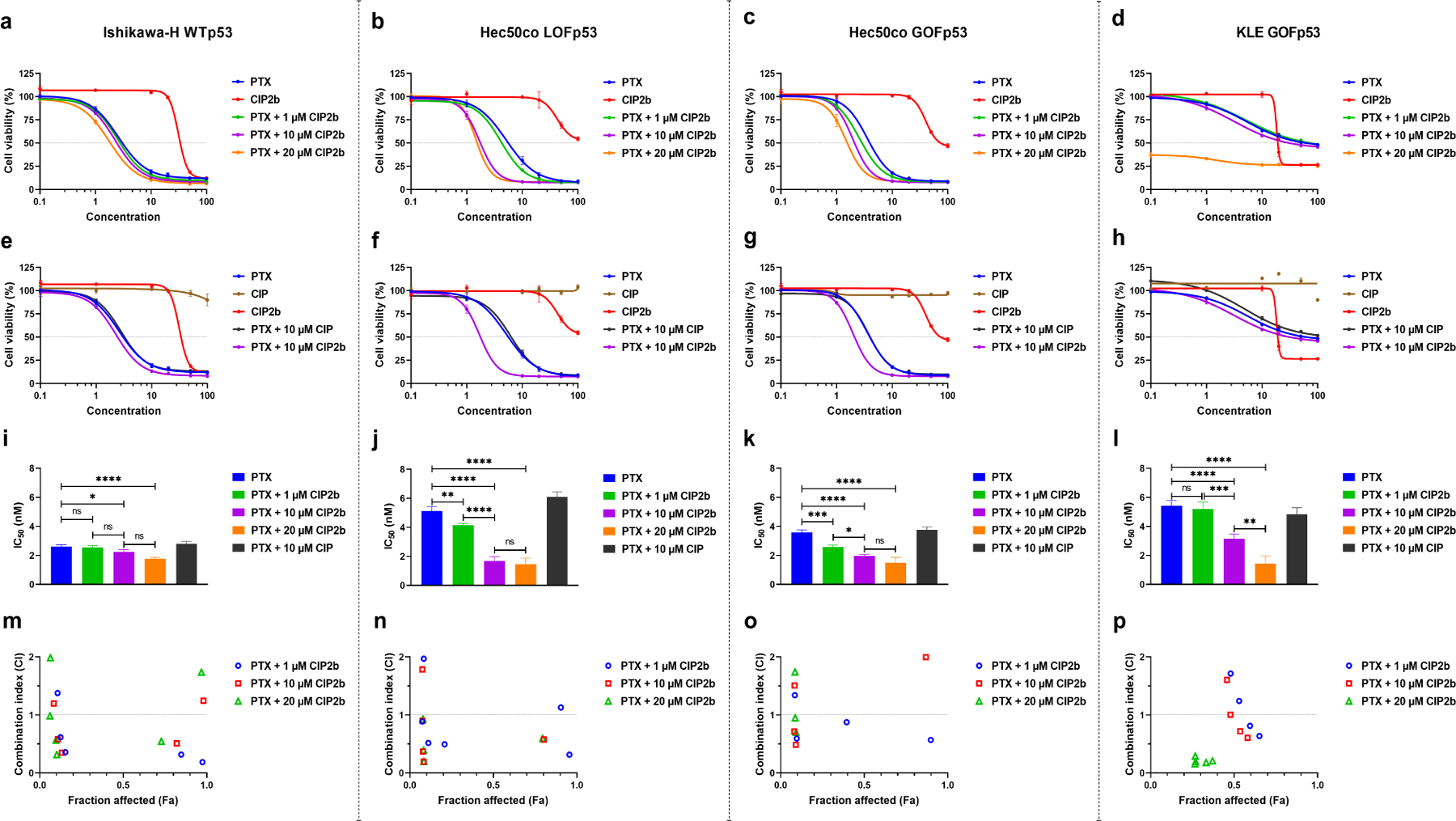
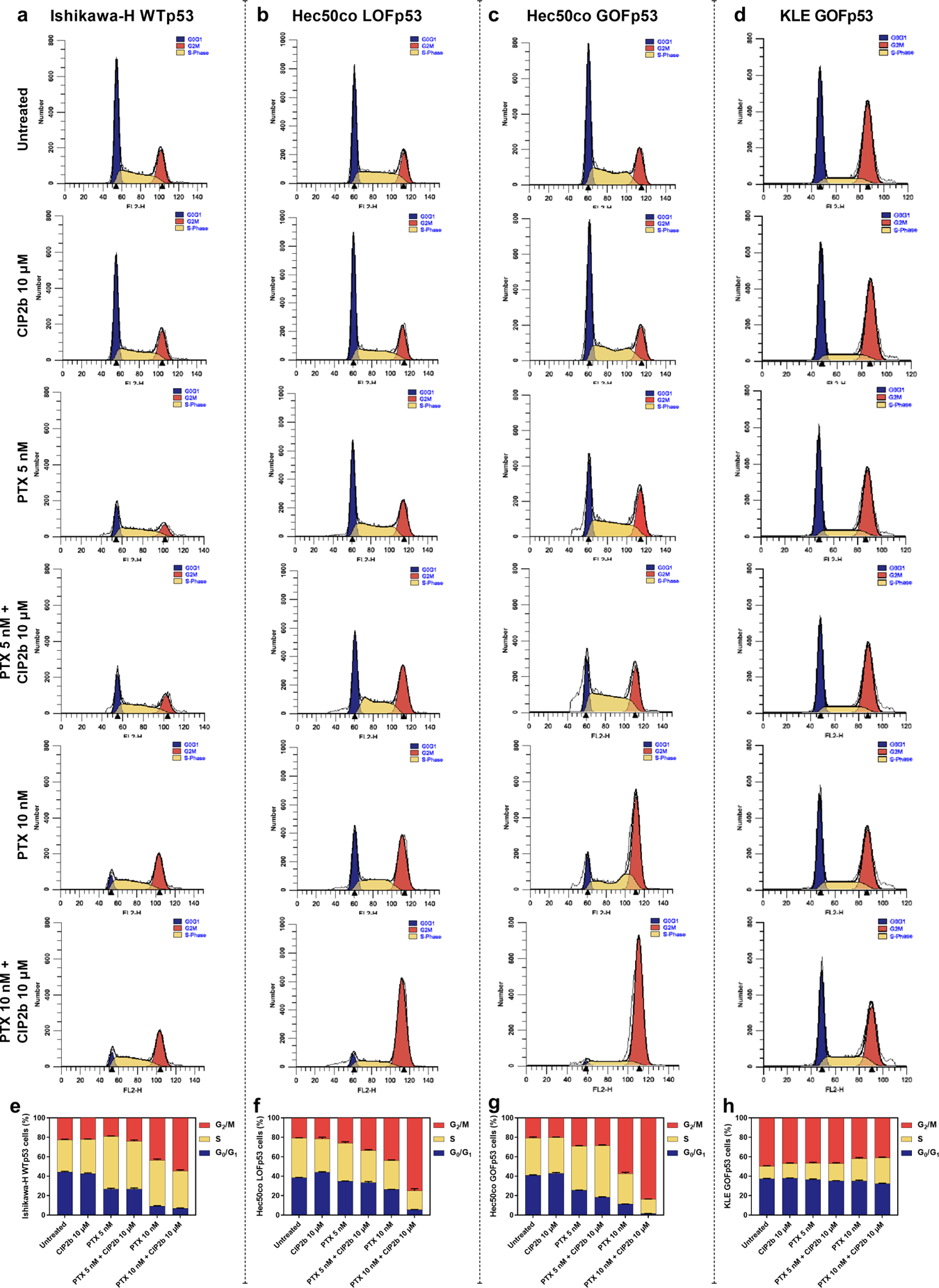
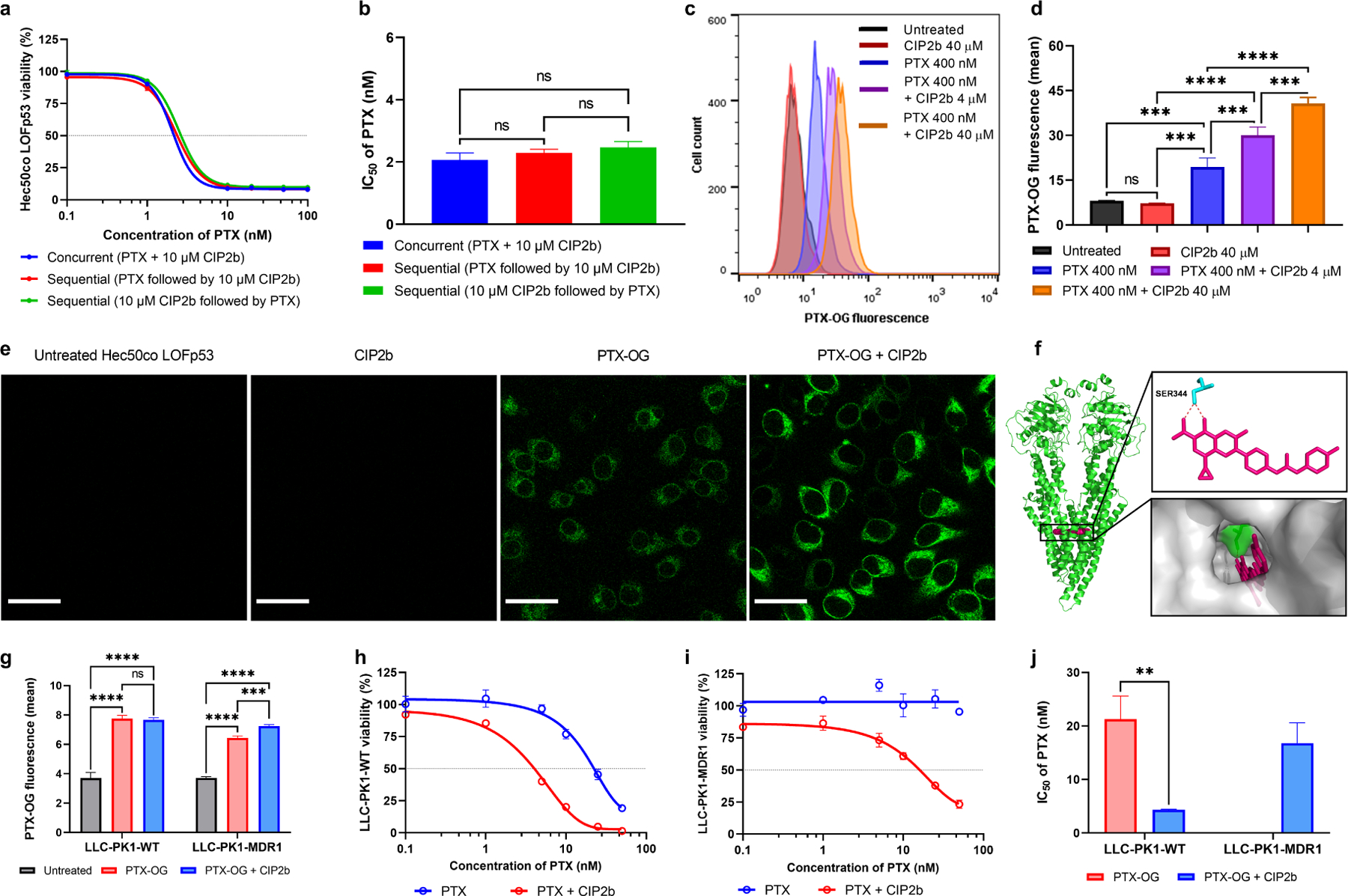
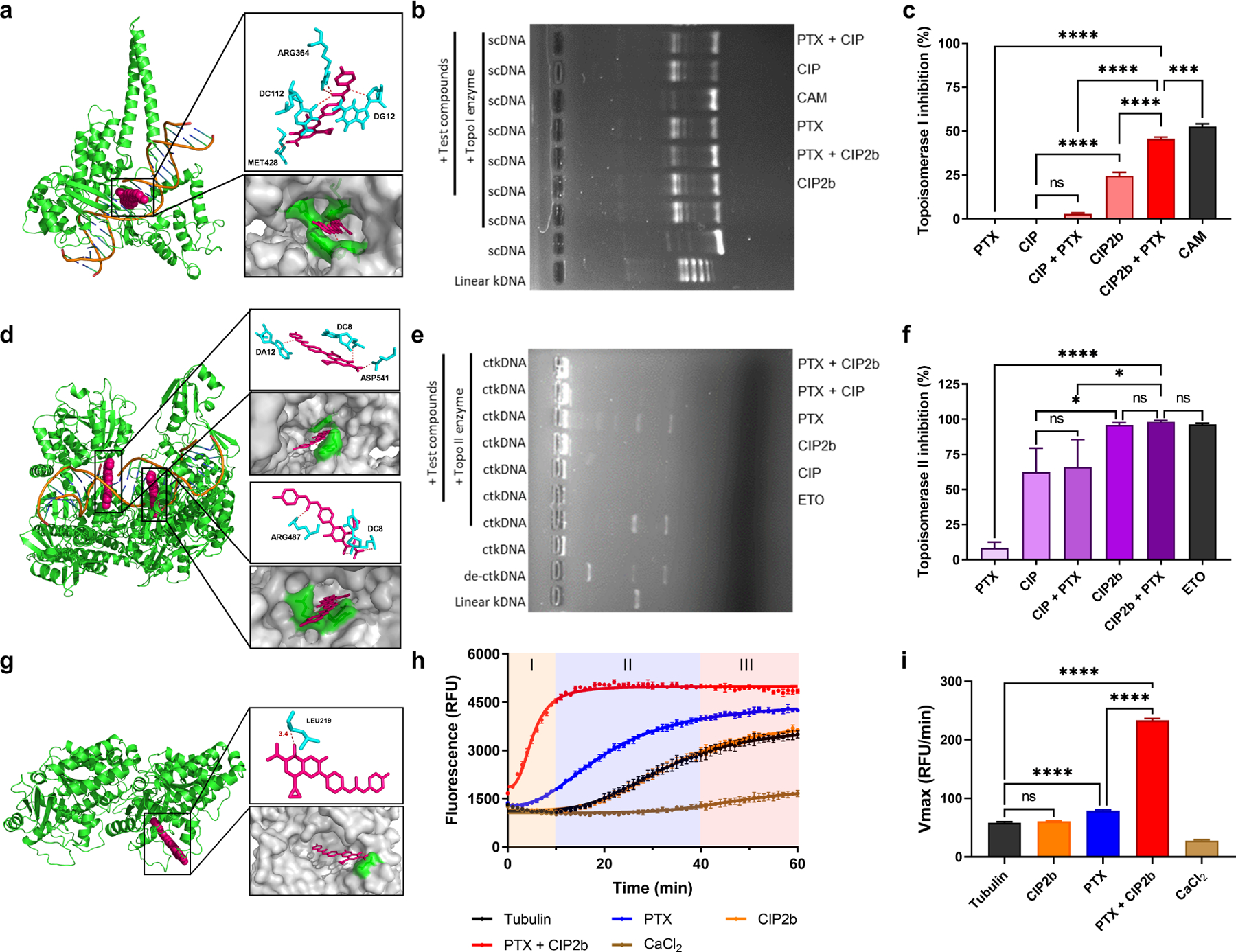
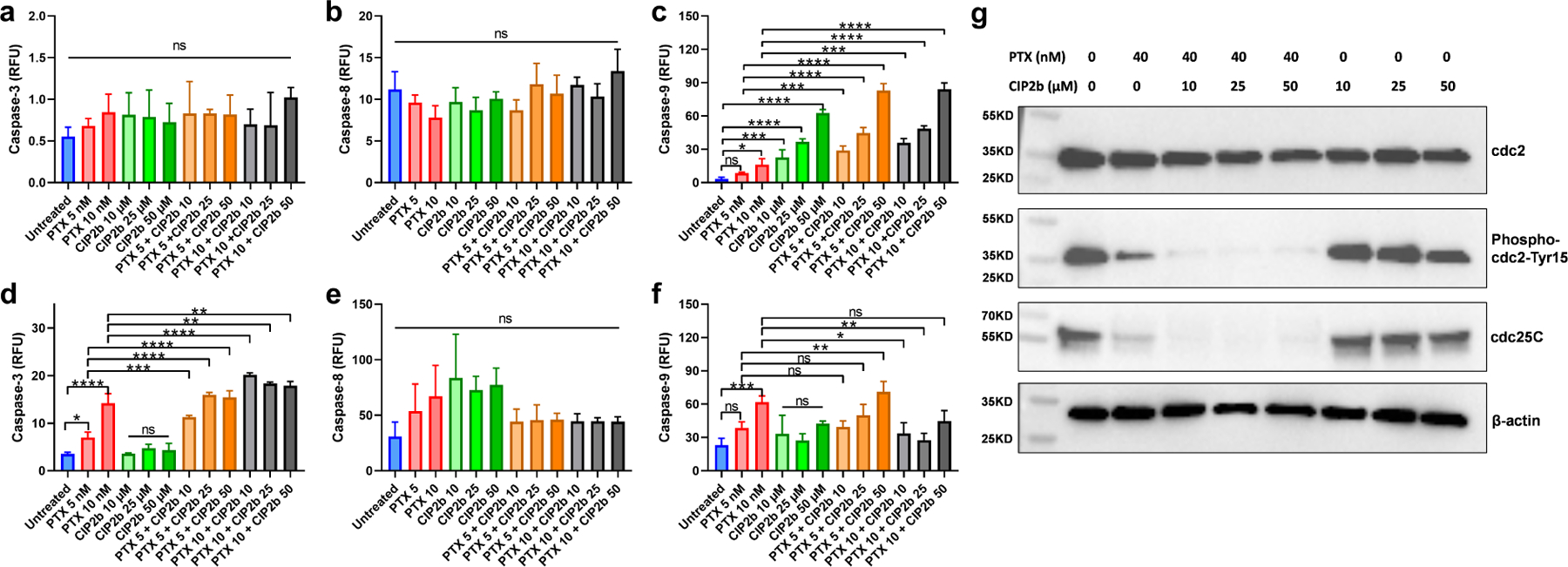
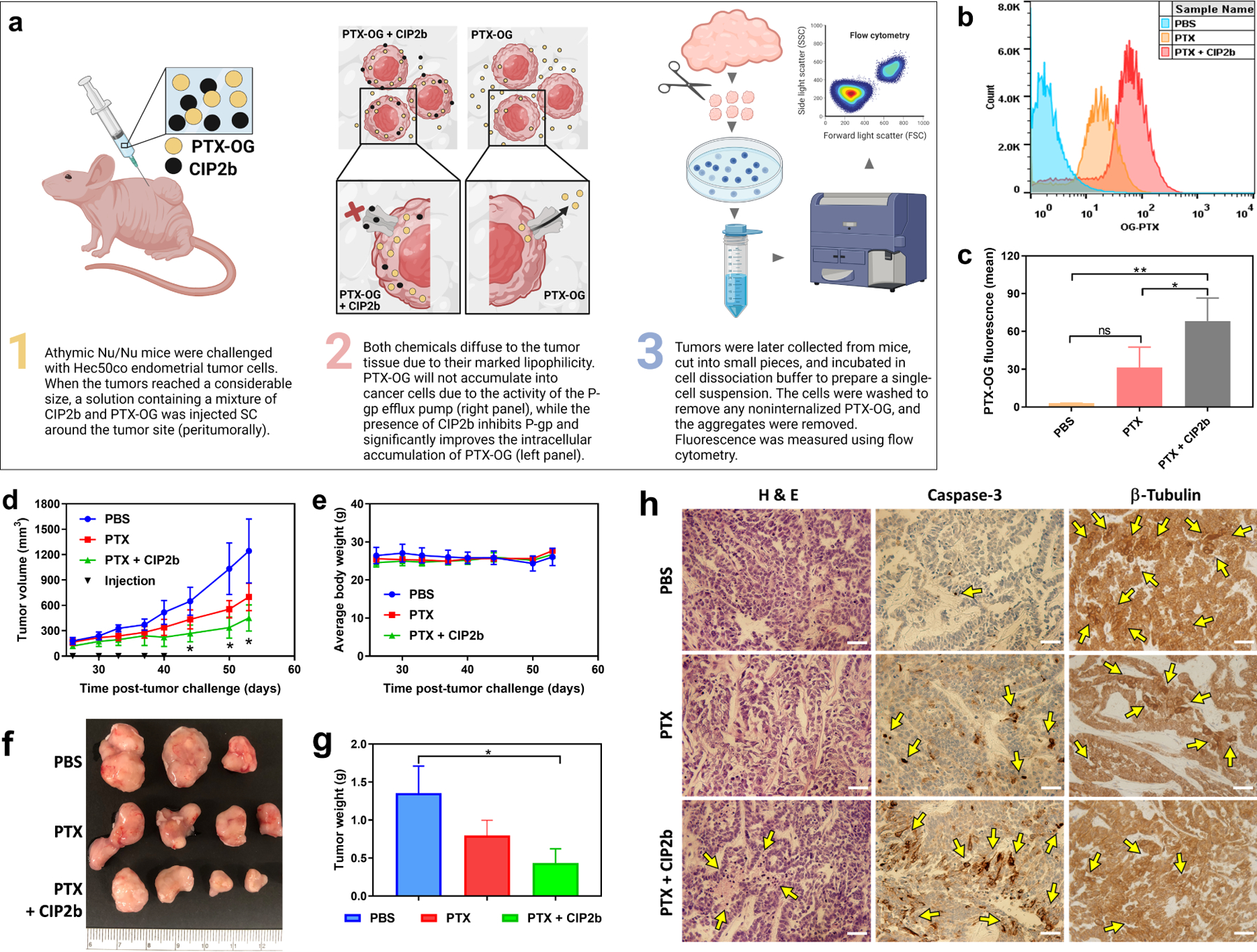
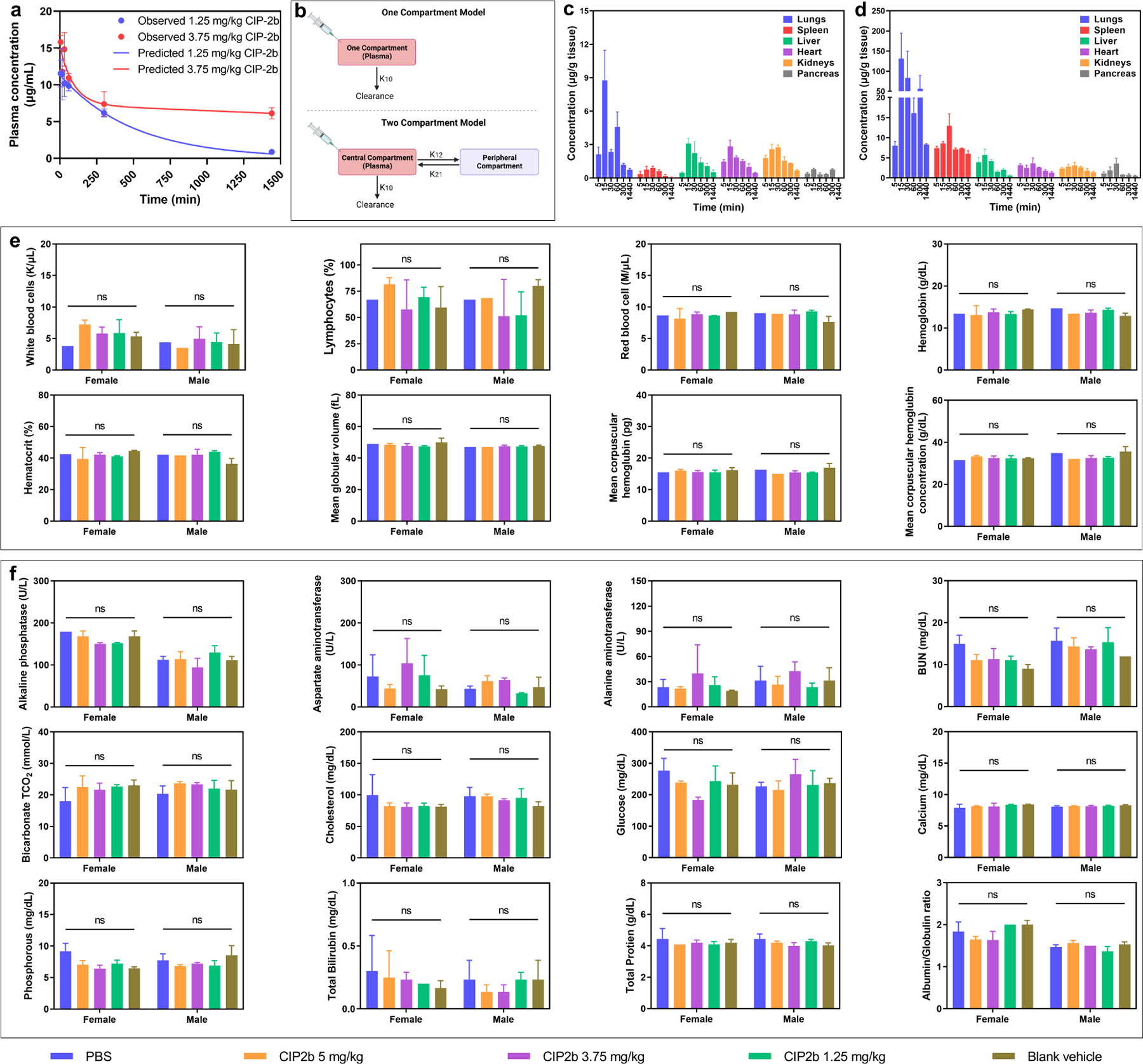
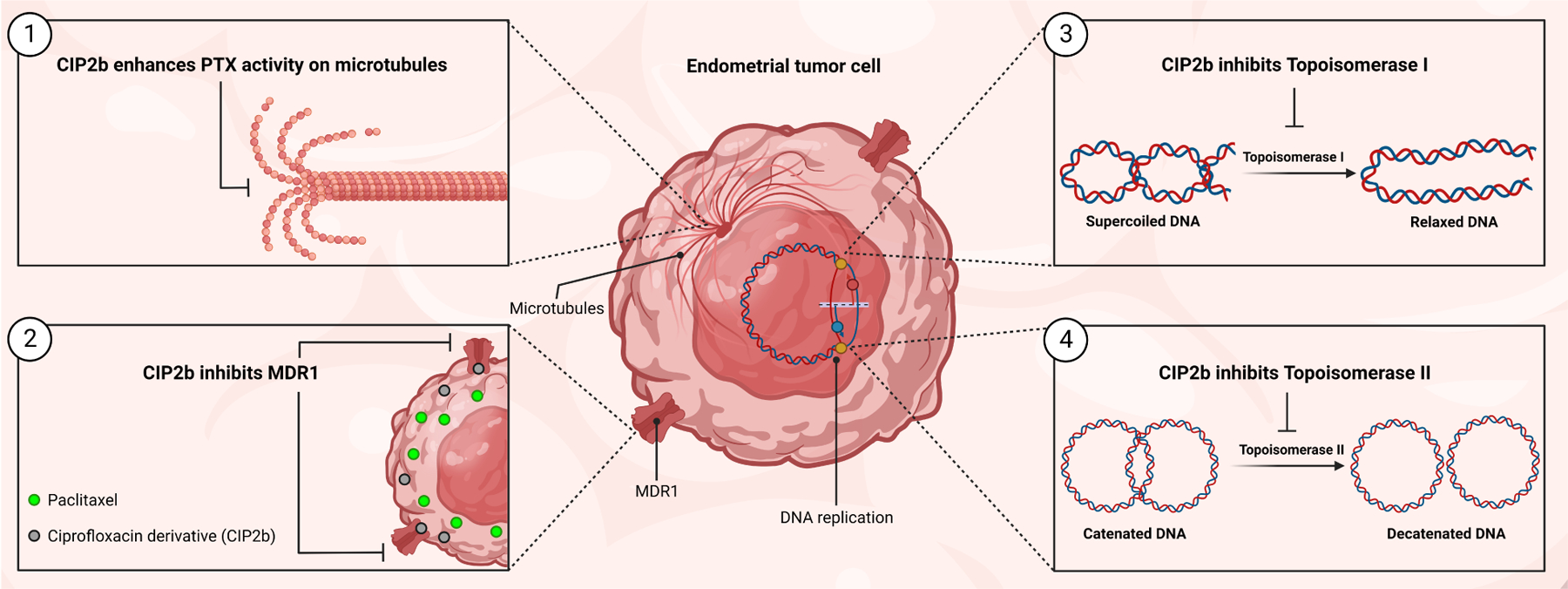
Dysfunction of the p53 gene and the presence of the MDR1 gene are associated with many malignant tumors including endometrial cancer and are responsible for cancer therapeutic resistance and poor survival. Thus, there is a critical need to devise novel combinatorial therapies with multiple mechanisms of action to overcome drug resistance. Here, we report a new ciprofloxacin derivative (CIP2b) tested either alone or in combination with taxanes against four human endometrial cancer cell lines. In vitro studies revealed that a combination of paclitaxel + CIP2b had synergistic cytotoxic effects against MDR1-expressing type-II human endometrial cancer cells with loss-of-function p53 (Hec50co LOFp53). Enhanced antitumor effects were confirmed by substantial increases in caspase-3 expression, cell population shifts toward the G2/M phase, and reduction of cdc2 phosphorylation. It was found that CIP2b targets multiple pathways including the inhibition of MDR1, topoisomerase I, and topoisomerase II, as well as enhancing the effects of paclitaxel (PTX) on microtubule assembly. In vivo treatment with the combination of PTX + CIP2b also led to significantly increased accumulation of PTX in tumors (compared to CIP2b alone) and reduction in tumor growth. Enhanced in vivo cytotoxic effects were confirmed by histological and immunohistochemical examination of the tumor tissues. Complete blood count and blood biochemistry data confirmed the absence of any apparent off-target toxicity. Thus, combination therapy involving PTX and CIP2b targeted multiple pathways and represents an approach that could result in improved tolerance and efficacy in patients with type-II endometrial cancer harboring the MDR1 gene and p53 mutations.
Keywords: Drug accumulation; Drug resistance; MDR1 and p53 genes; Microtubules; Molecular combinatorial therapy; Topoisomerases.
Copyright © 2023 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Mota A, Oltra SS, Selenica P, Moiola CP, Casas–Arozamena C, Lopez–Gil C, Diaz E, Gatius S, Ruiz–Miro M, Calvo A, Rojo–Sebastian A, Hurtado P, Pineiro R, Colas E, Gil–Moreno A, Reis–Filho JS, Muinelo–Romay L, Abal M, Matias–Guiu X, Weigelt B, Moreno–Bueno G, Intratumor genetic heterogeneity and clonal evolution to decode endometrial cancer progression, Oncogene 41(13) (2022) 1835–1850. - PMC - PubMed
-
- Gatius S, Cuevas D, Fernandez C, Roman–Canal B, Adamoli V, Piulats JM, Eritja N, Martin-Satue M, Moreno–Bueno G, Matias–Guiu X, Tumor Heterogeneity in Endometrial Carcinoma: Practical Consequences, Pathobiology 85(1–2) (2018) 35–40. - PubMed
-
- Setiawan VW, Yang HP, Pike MC, McCann SE, Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, van den Brandt PA, van de Vijver K, Thompson PJ, Australian G National Endometrial Cancer Study, Strom BL, Spurdle AB, Soslow RA, Shu XO, Schairer C, Sacerdote C, Rohan TE, Robien K, Risch HA, Ricceri F, Rebbeck TR, Rastogi R, Prescott J, Polidoro S, Park Y, Olson SH, Moysich KB, Miller AB, McCullough ML, Matsuno RK, Magliocco AM, Lurie G, Lu L, Lissowska J, Liang X, Lacey JV Jr., Kolonel LN, Henderson BE, Hankinson SE, Hakansson N, Goodman MT, Gaudet MM, Garcia–Closas M, Friedenreich CM, Freudenheim JL, Doherty J, De Vivo I, Courneya KS, Cook LS, Chen C, Cerhan JR, Cai H, Brinton LA, Bernstein L, Anderson KE, Anton-Culver H, Schouten LJ, Horn–Ross PL, Type I and II endometrial cancers: have they different risk factors?, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 31(20) (2013) 2607–18. - PMC - PubMed
-
- Murali R, Soslow RA, Weigelt B, Classification of endometrial carcinoma: more than two types, The Lancet. Oncology 15(7) (2014) e268–78. - PubMed
-
- Lu KH, Broaddus RR, Endometrial Cancer, The New England journal of medicine 383(21) (2020) 2053–2064. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
