

U1 snRNP increases RNA Pol II elongation rate to enable synthesis of long genes
- PMID: 36965480
- PMCID: PMC10135401
- DOI: 10.1016/j.molcel.2023.03.002
U1 snRNP increases RNA Pol II elongation rate to enable synthesis of long genes
Abstract
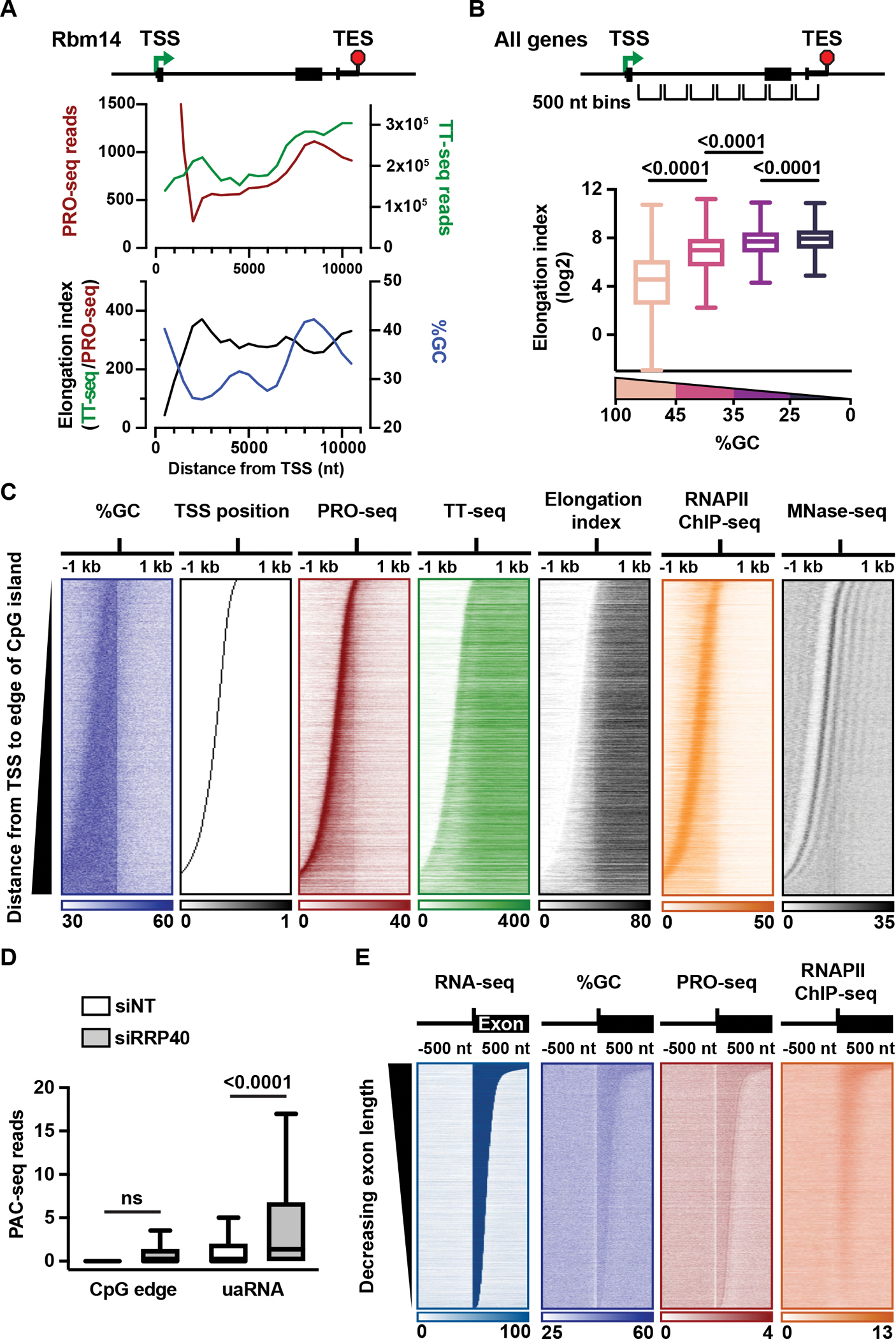
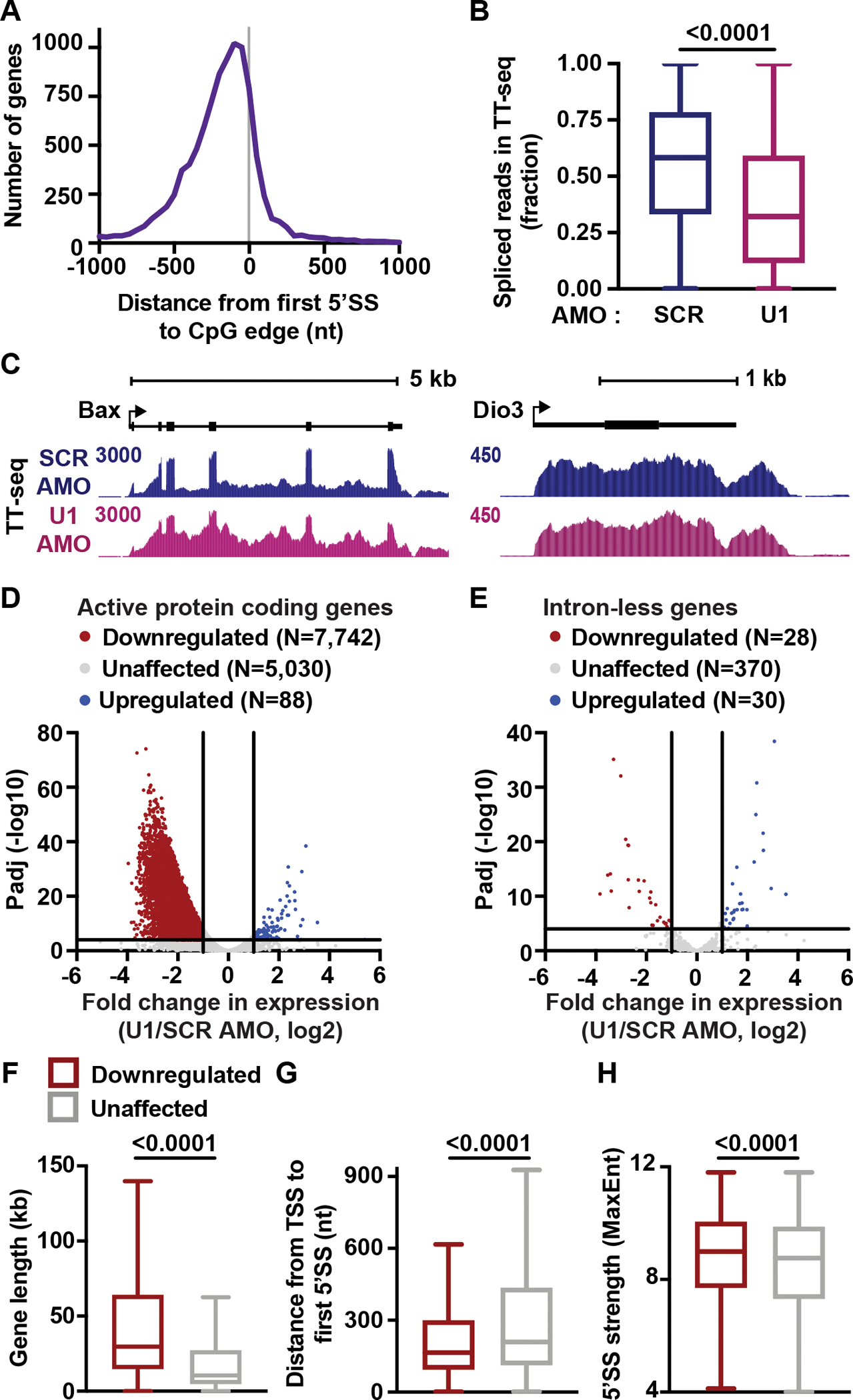
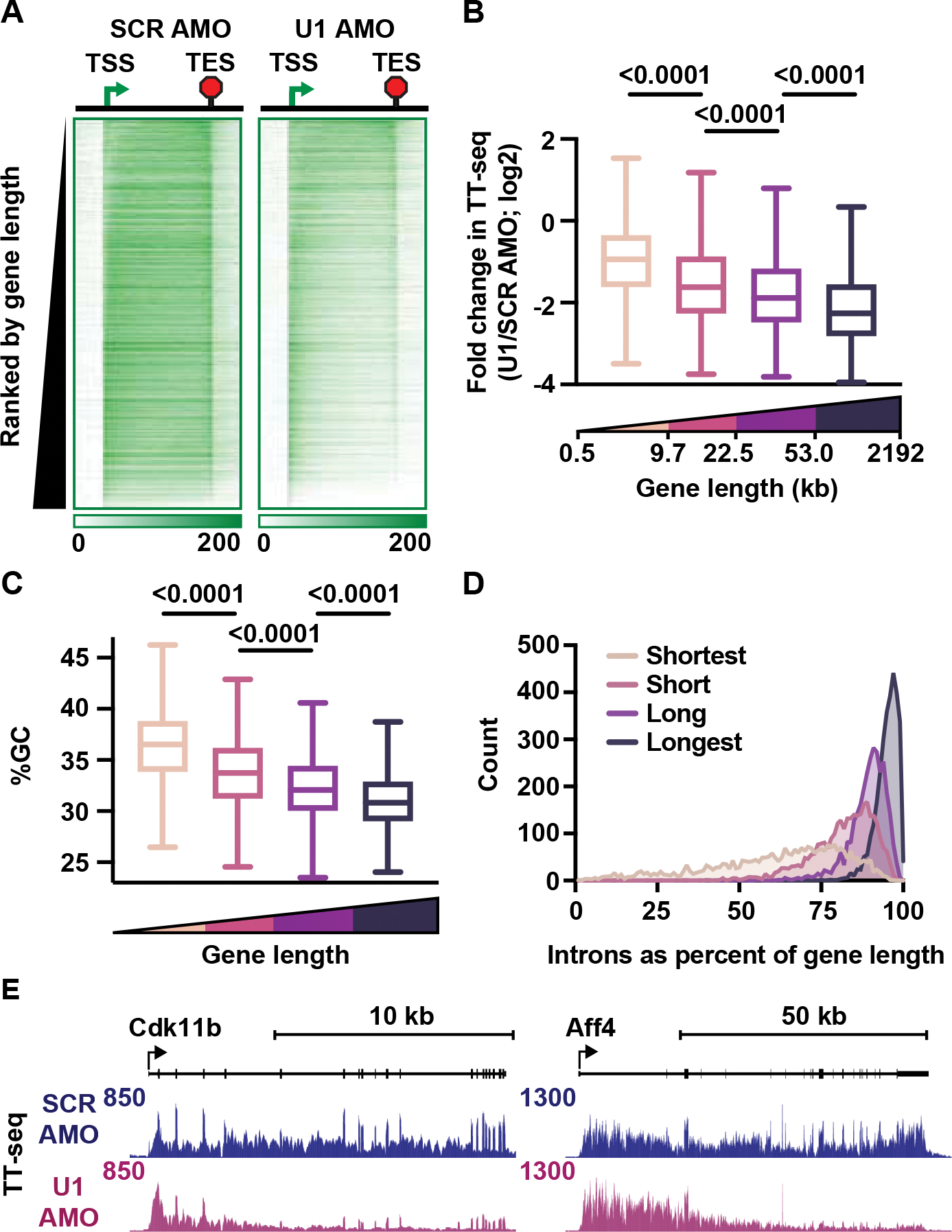
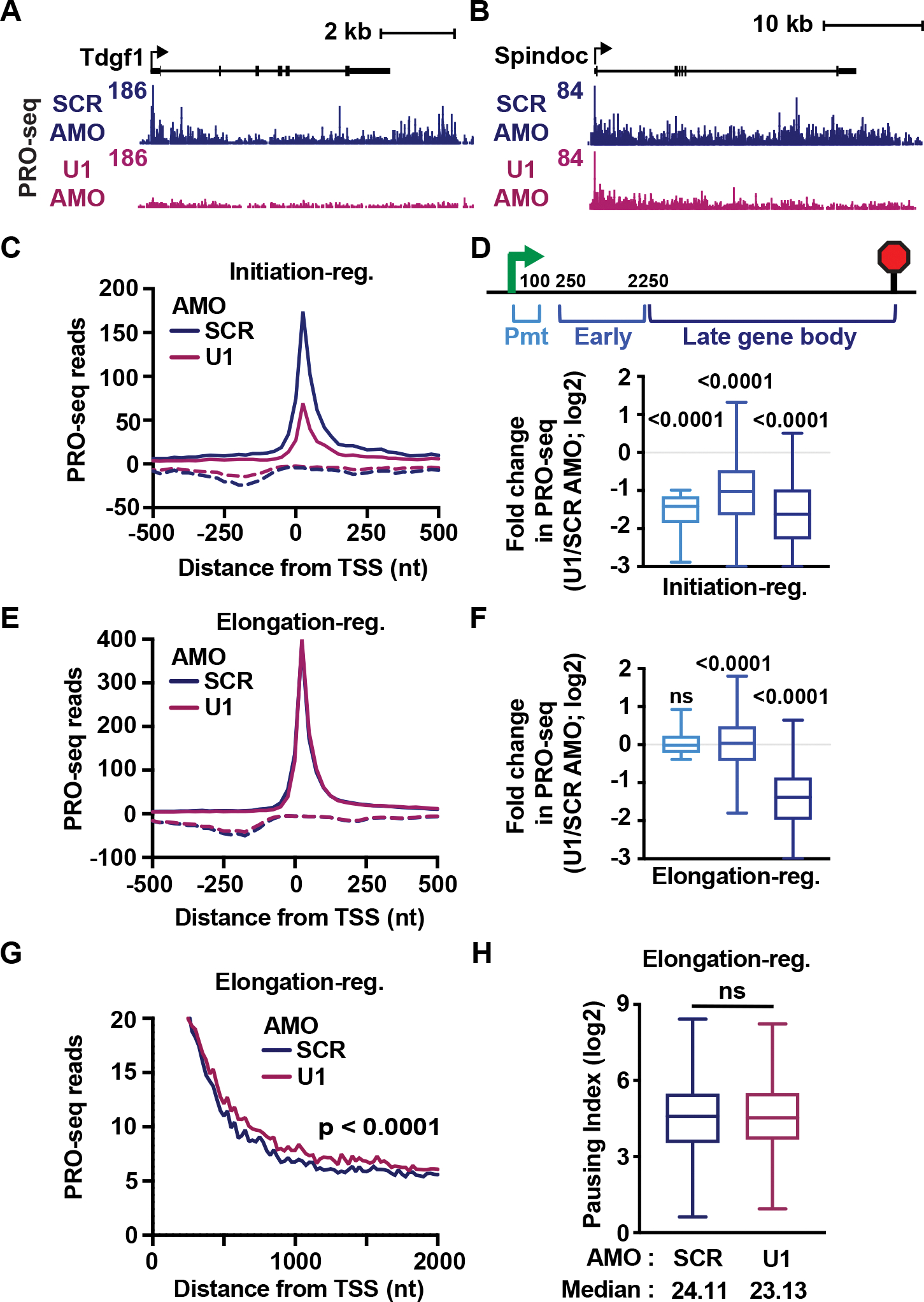
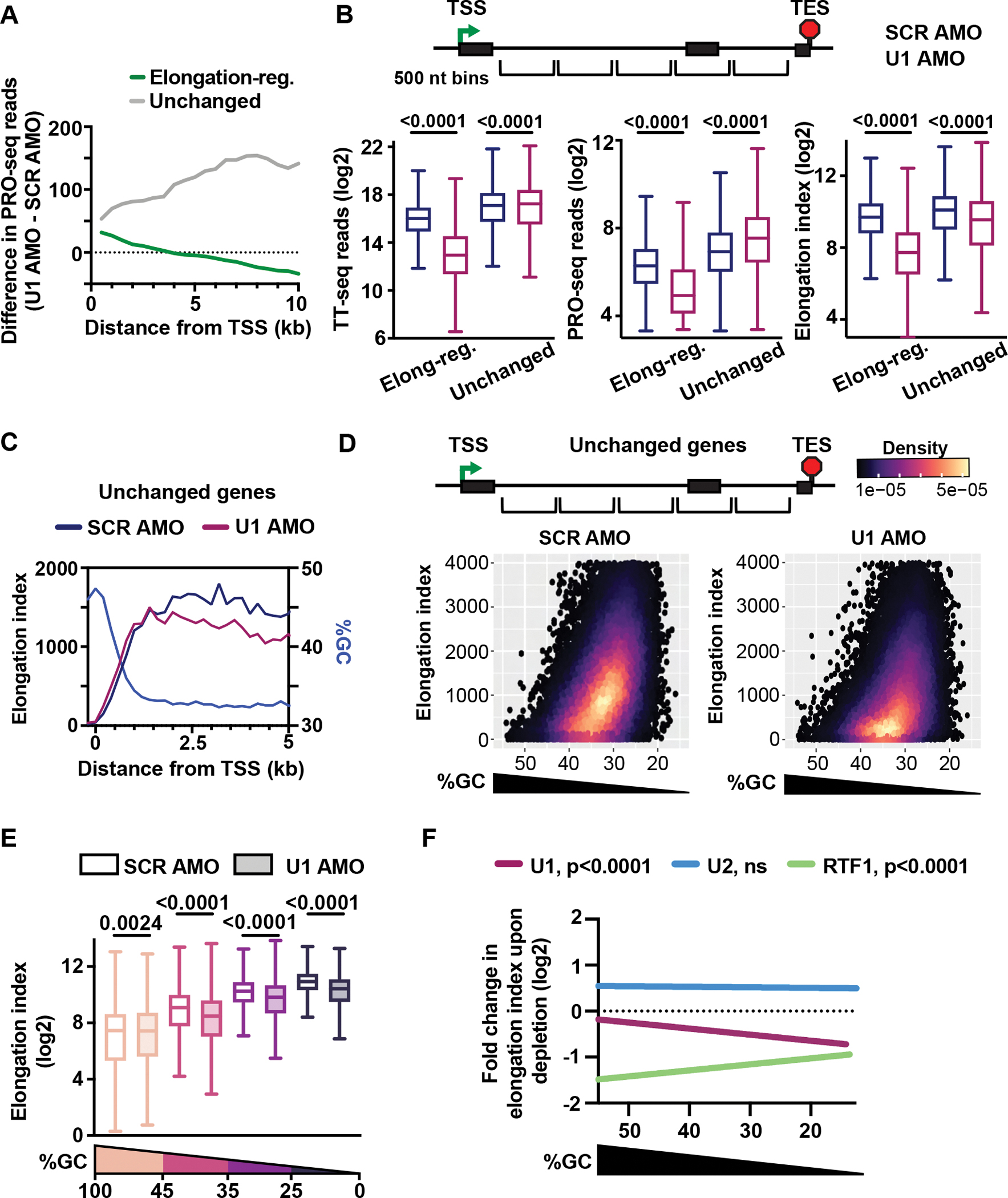
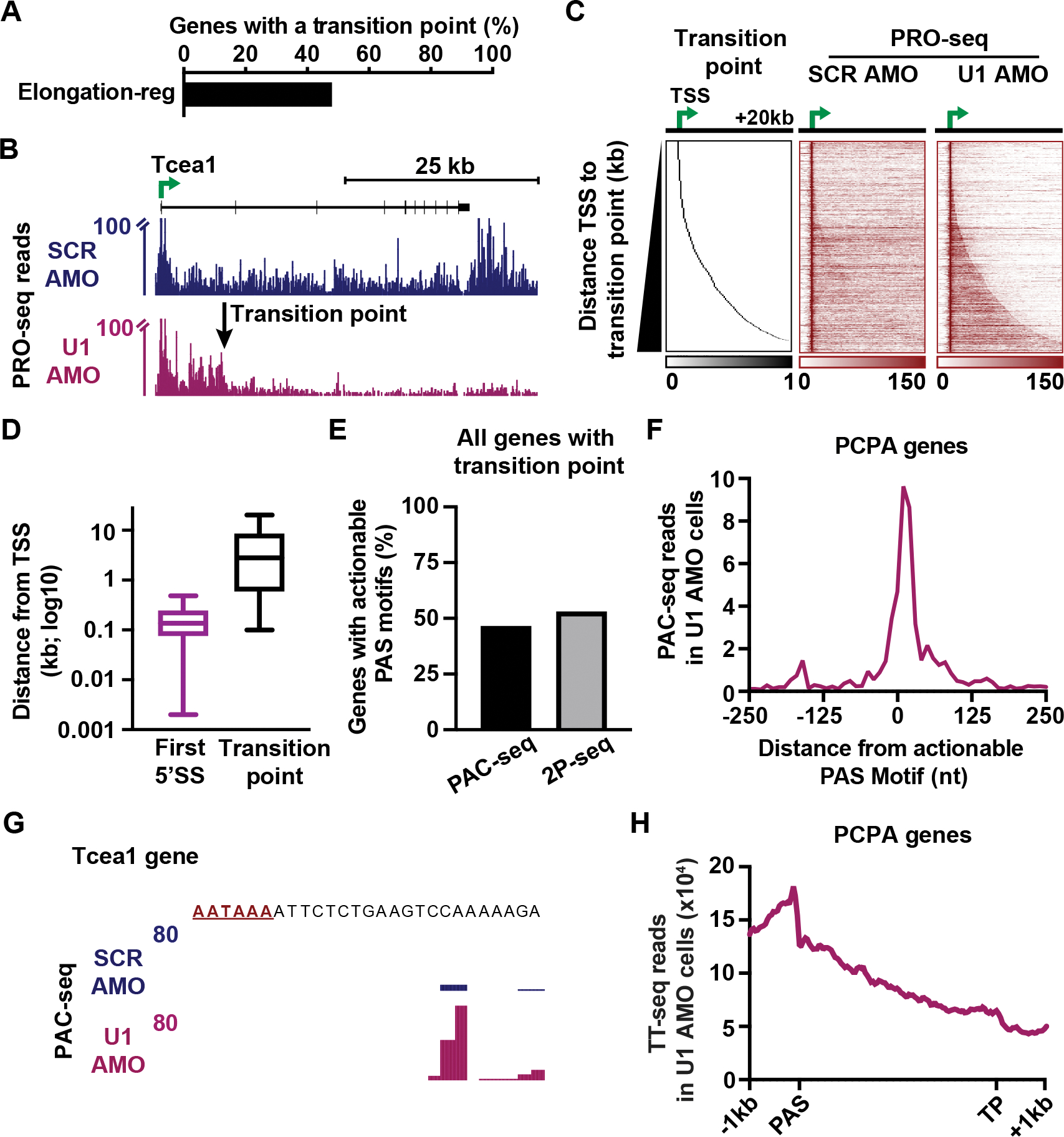
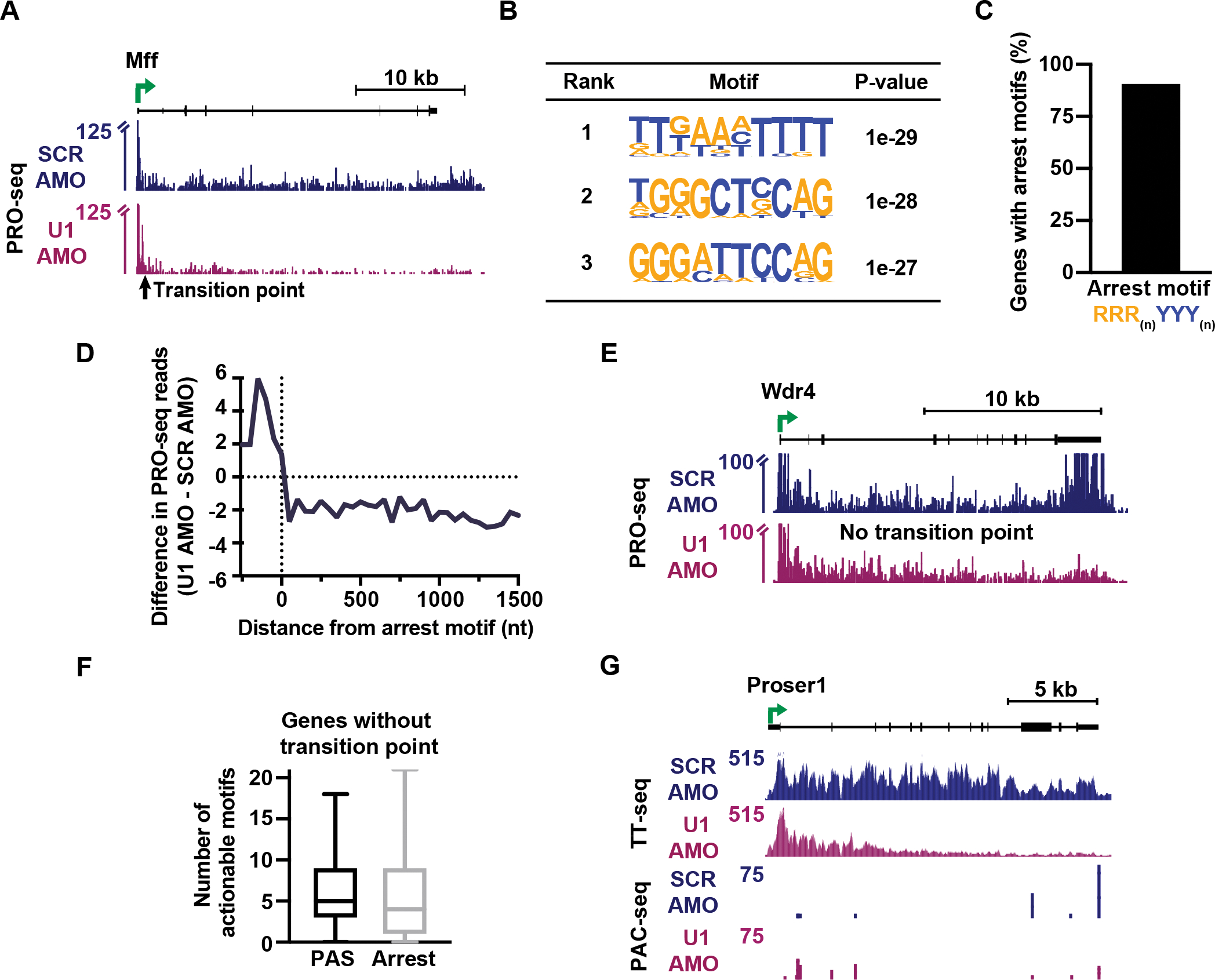
The expansion of introns within mammalian genomes poses a challenge for the production of full-length messenger RNAs (mRNAs), with increasing evidence that these long AT-rich sequences present obstacles to transcription. Here, we investigate RNA polymerase II (RNAPII) elongation at high resolution in mammalian cells and demonstrate that RNAPII transcribes faster across introns. Moreover, we find that this acceleration requires the association of U1 snRNP (U1) with the elongation complex at 5' splice sites. The role of U1 to stimulate elongation rate through introns reduces the frequency of both premature termination and transcriptional arrest, thereby dramatically increasing RNA production. We further show that changes in RNAPII elongation rate due to AT content and U1 binding explain previous reports of pausing or termination at splice junctions and the edge of CpG islands. We propose that U1-mediated acceleration of elongation has evolved to mitigate the risks that long AT-rich introns pose to transcript completion.
Keywords: CpG island; RNA polymerase II; U1 snRNP; co-transcriptional splicing; elongation factors; elongation rate; long genes; nascent RNA; sequence content; transcription regulation.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.A. received research funding from Novartis not related to this work, is on the SAB of CAMP4 Therapeutics, and is a member of the Advisory Board of Molecular Cell.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
