

Integrative proteogenomic characterization of early esophageal cancer
- PMID: 36966136
- PMCID: PMC10039899
- DOI: 10.1038/s41467-023-37440-w
Integrative proteogenomic characterization of early esophageal cancer
Abstract
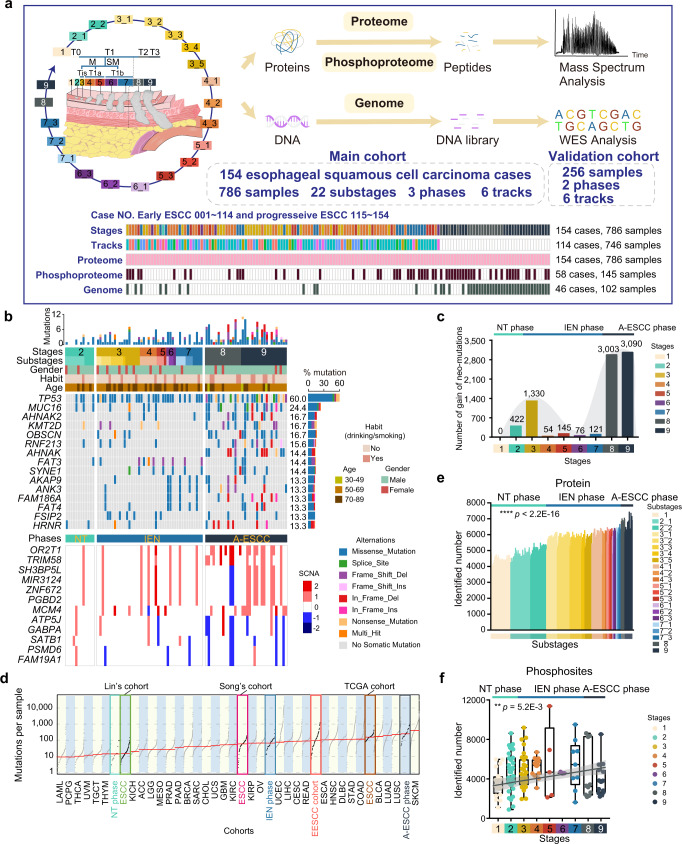
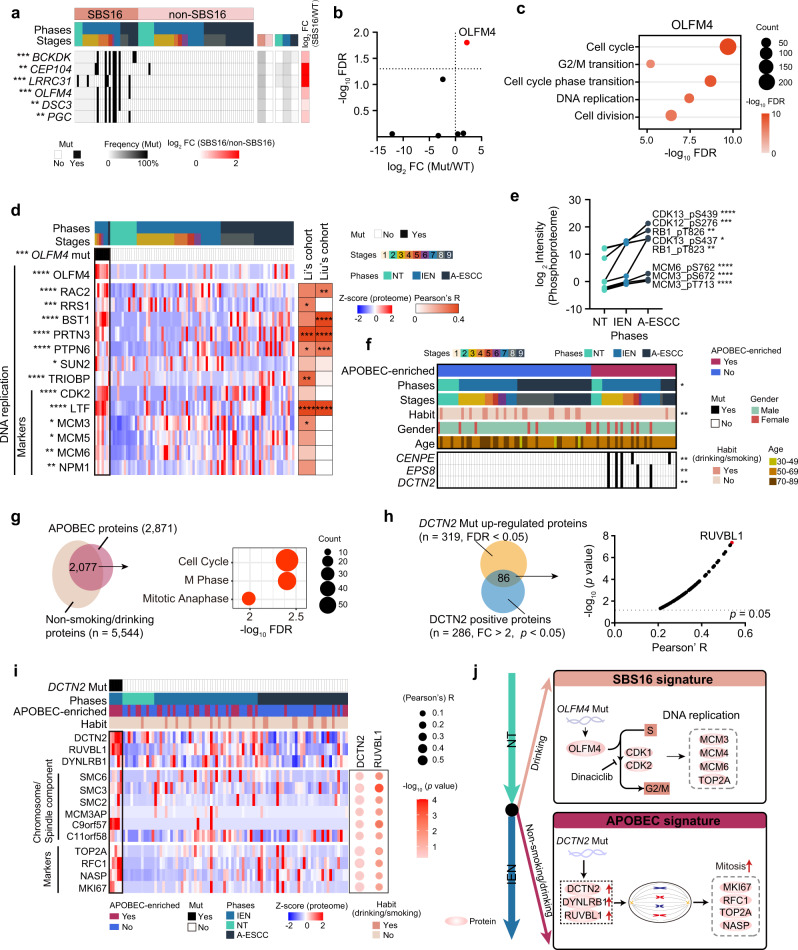
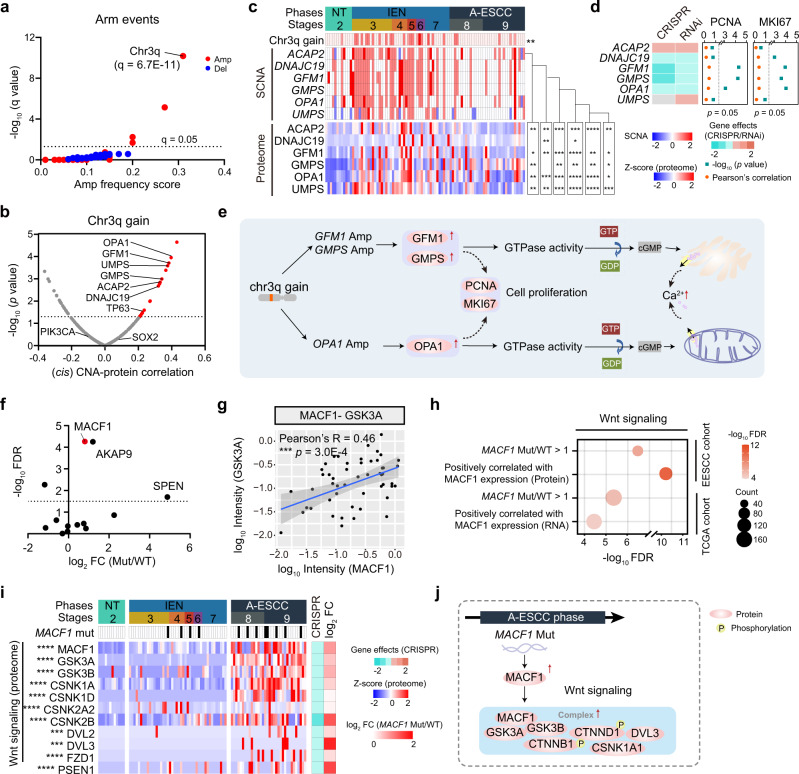
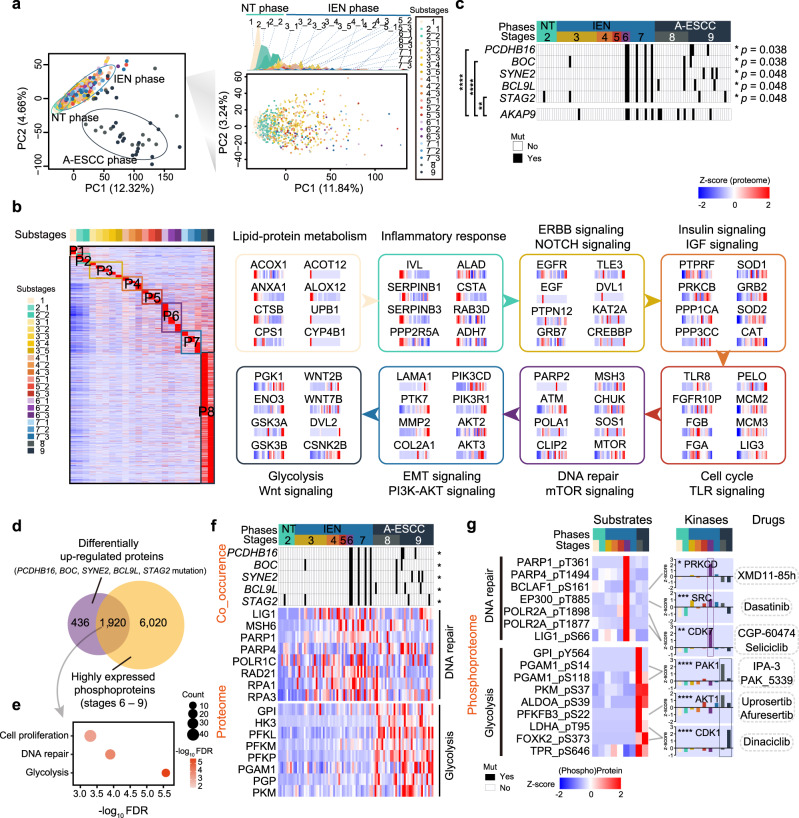
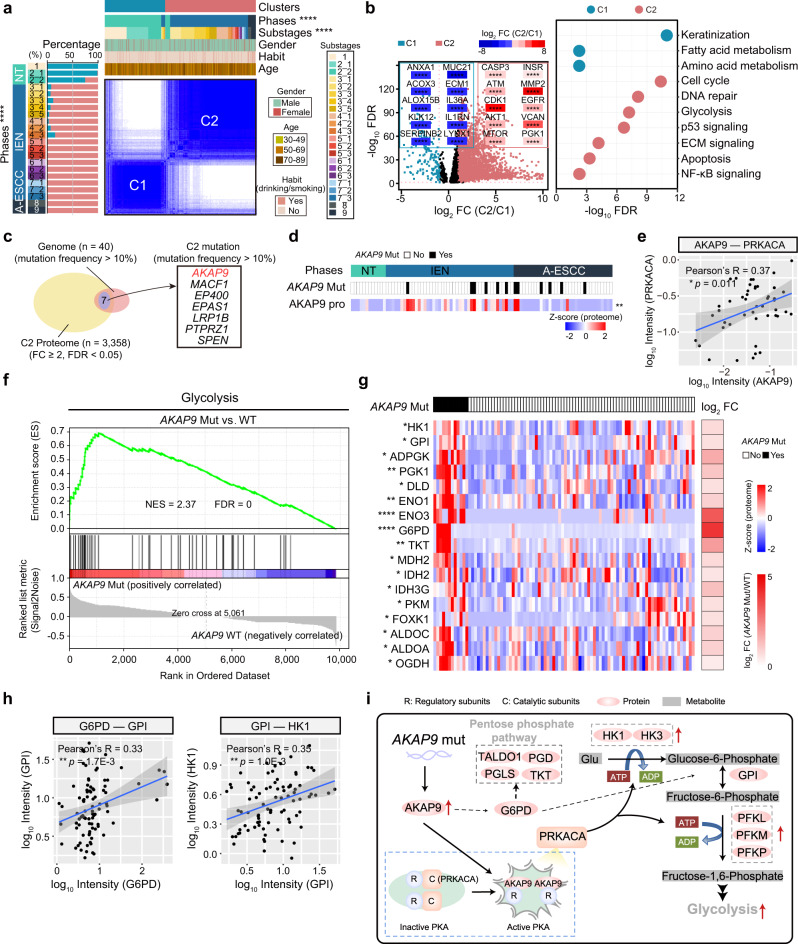
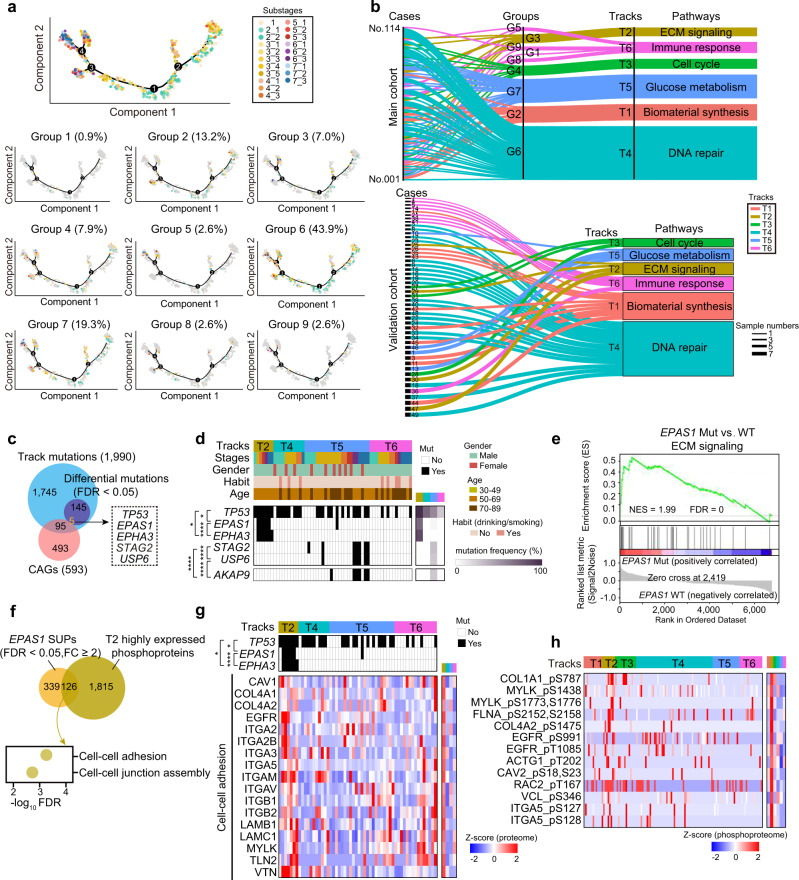
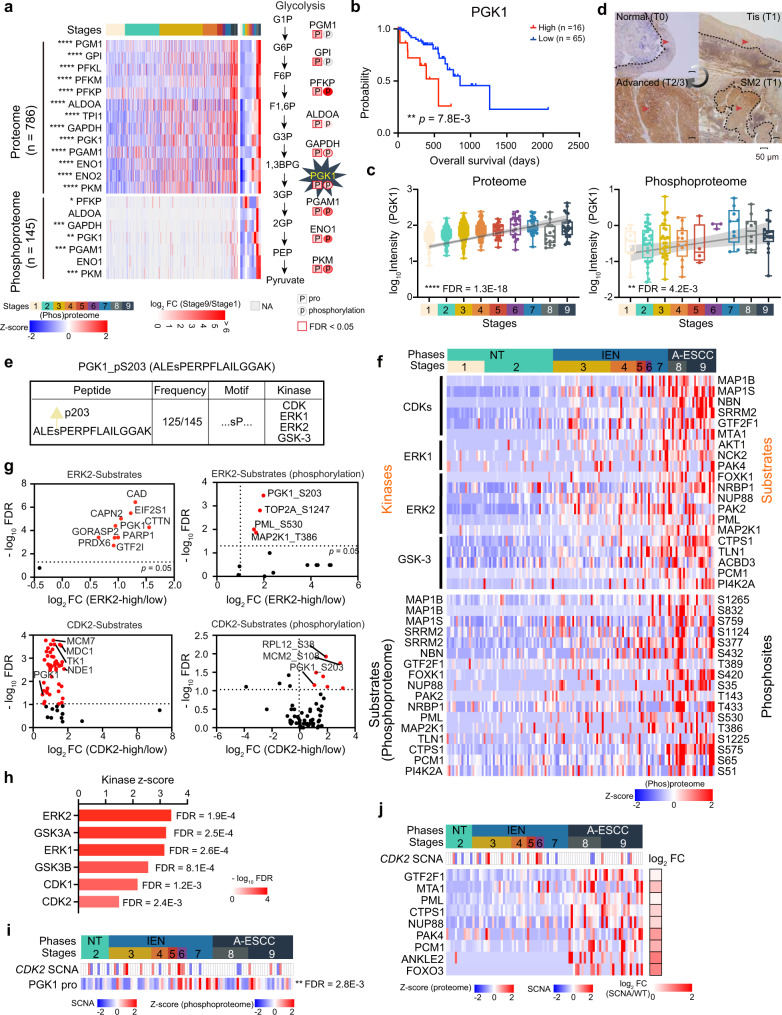
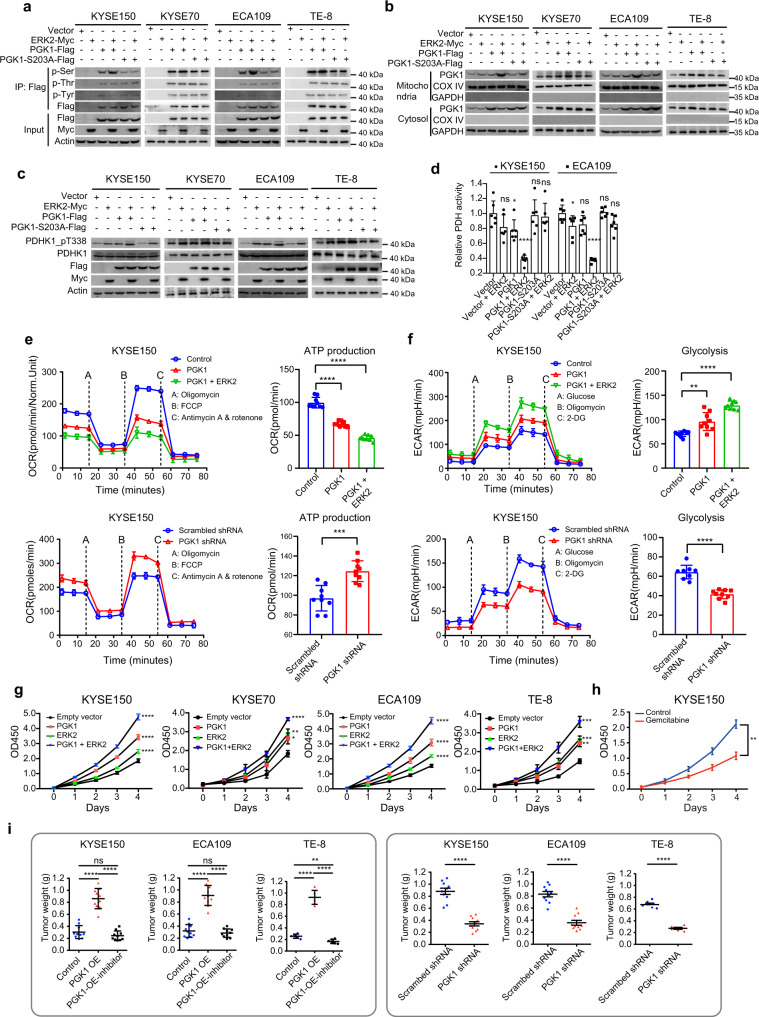
Esophageal squamous cell carcinoma (ESCC) is malignant while the carcinogenesis is still unclear. Here, we perform a comprehensive multi-omics analysis of 786 trace-tumor-samples from 154 ESCC patients, covering 9 histopathological stages and 3 phases. Proteogenomics elucidates cancer-driving waves in ESCC progression, and reveals the molecular characterization of alcohol drinking habit associated signatures. We discover chromosome 3q gain functions in the transmit from nontumor to intraepithelial neoplasia phases, and find TP53 mutation enhances DNA replication in intraepithelial neoplasia phase. The mutations of AKAP9 and MCAF1 upregulate glycolysis and Wnt signaling, respectively, in advanced-stage ESCC phase. Six major tracks related to different clinical features during ESCC progression are identified, which is validated by an independent cohort with another 256 samples. Hyperphosphorylated phosphoglycerate kinase 1 (PGK1, S203) is considered as a drug target in ESCC progression. This study provides insight into the understanding of ESCC molecular mechanism and the development of therapeutic targets.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
