

Alzheimer's disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression
- PMID: 36970154
- PMCID: PMC10033629
- DOI: 10.3389/fnsyn.2023.1129036
Alzheimer's disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression
Abstract
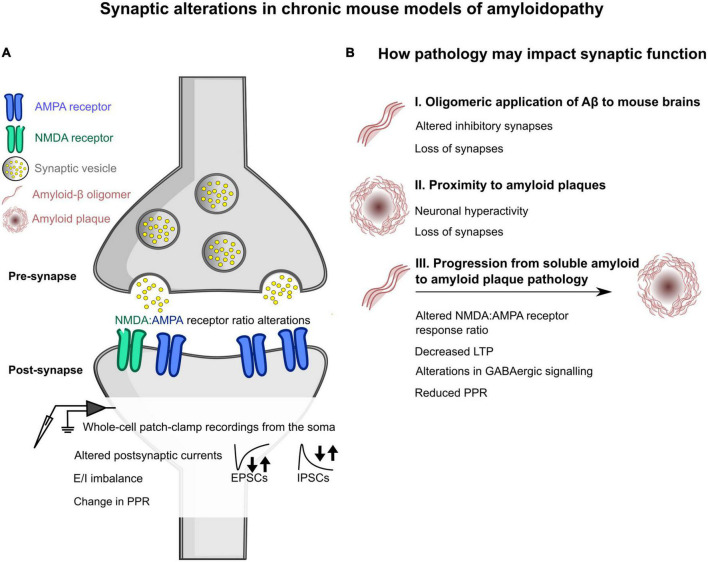
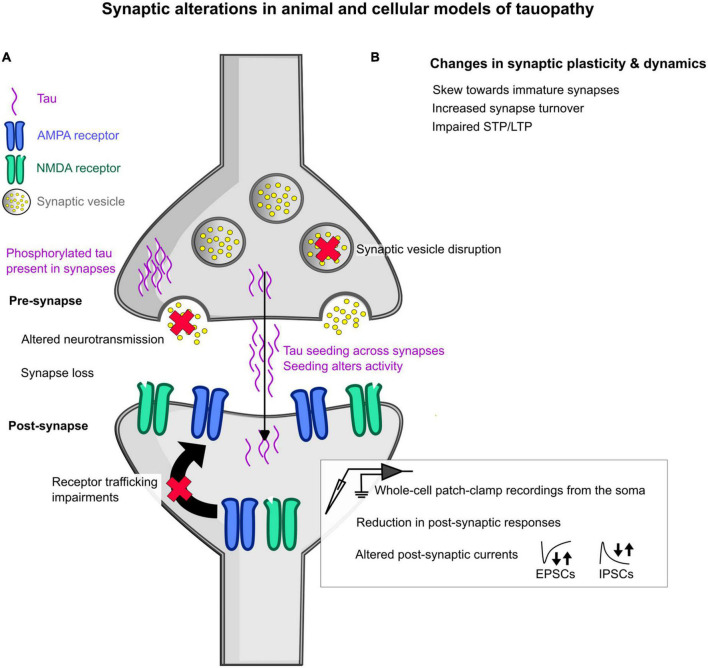
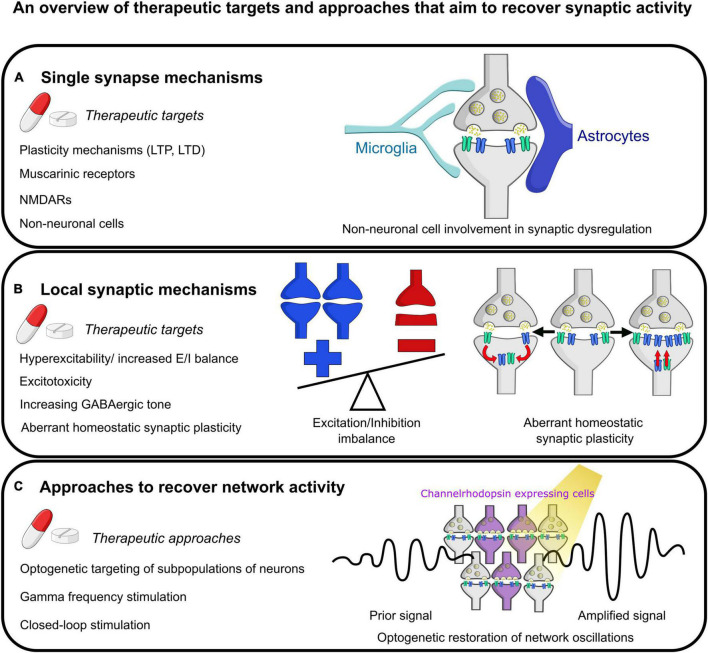
The synapse has consistently been considered a vulnerable and critical target within Alzheimer's disease, and synapse loss is, to date, one of the main biological correlates of cognitive decline within Alzheimer's disease. This occurs prior to neuronal loss with ample evidence that synaptic dysfunction precedes this, in support of the idea that synaptic failure is a crucial stage within disease pathogenesis. The two main pathological hallmarks of Alzheimer's disease, abnormal aggregates of amyloid or tau proteins, have had demonstrable effects on synaptic physiology in animal and cellular models of Alzheimer's disease. There is also growing evidence that these two proteins may have a synergistic effect on neurophysiological dysfunction. Here, we review some of the main findings of synaptic alterations in Alzheimer's disease, and what we know from Alzheimer's disease animal and cellular models. First, we briefly summarize some of the human evidence to suggest that synapses are altered, including how this relates to network activity. Subsequently, animal and cellular models of Alzheimer's disease are considered, highlighting mouse models of amyloid and tau pathology and the role these proteins may play in synaptic dysfunction, either in isolation or examining how the two pathologies may interact in dysfunction. This specifically focuses on neurophysiological function and dysfunction observed within these animal models, typically measured using electrophysiology or calcium imaging. Following synaptic dysfunction and loss, it would be impossible to imagine that this would not alter oscillatory activity within the brain. Therefore, this review also discusses how this may underpin some of the aberrant oscillatory patterns seen in animal models of Alzheimer's disease and human patients. Finally, an overview of some key directions and considerations in the field of synaptic dysfunction in Alzheimer's disease is covered. This includes current therapeutics that are targeted specifically at synaptic dysfunction, but also methods that modulate activity to rescue aberrant oscillatory patterns. Other important future avenues of note in this field include the role of non-neuronal cell types such as astrocytes and microglia, and mechanisms of dysfunction independent of amyloid and tau in Alzheimer's disease. The synapse will certainly continue to be an important target within Alzheimer's disease for the foreseeable future.
Keywords: Alzheimer’s disease; amyloid; dysfunction; electrophysiology; oscillations; synapse; synaptic transmission; tau.
Copyright © 2023 Meftah and Gan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Ahmed Z., Cooper J., Murray T., Garn K., McNaughton E., Clarke H., et al. (2014). A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127 667–683. 10.1007/s00401-014-1254-6 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources