

ATM inhibition enhances the efficacy of radiation across distinct molecular subgroups of pediatric high-grade glioma
- PMID: 36971093
- PMCID: PMC10547515
- DOI: 10.1093/neuonc/noad064
ATM inhibition enhances the efficacy of radiation across distinct molecular subgroups of pediatric high-grade glioma
Abstract
Background: Pediatric high-grade glioma (pHGG) is largely incurable and accounts for most brain tumor-related deaths in children. Radiation is a standard therapy, yet the benefit from this treatment modality is transient, and most children succumb to disease within 2 years. Recent large-scale genomic studies suggest that pHGG has alterations in DNA damage response (DDR) pathways that induce resistance to DNA damaging agents. The aim of this study was to evaluate the therapeutic potential and molecular consequences of combining radiation with selective DDR inhibition in pHGG.
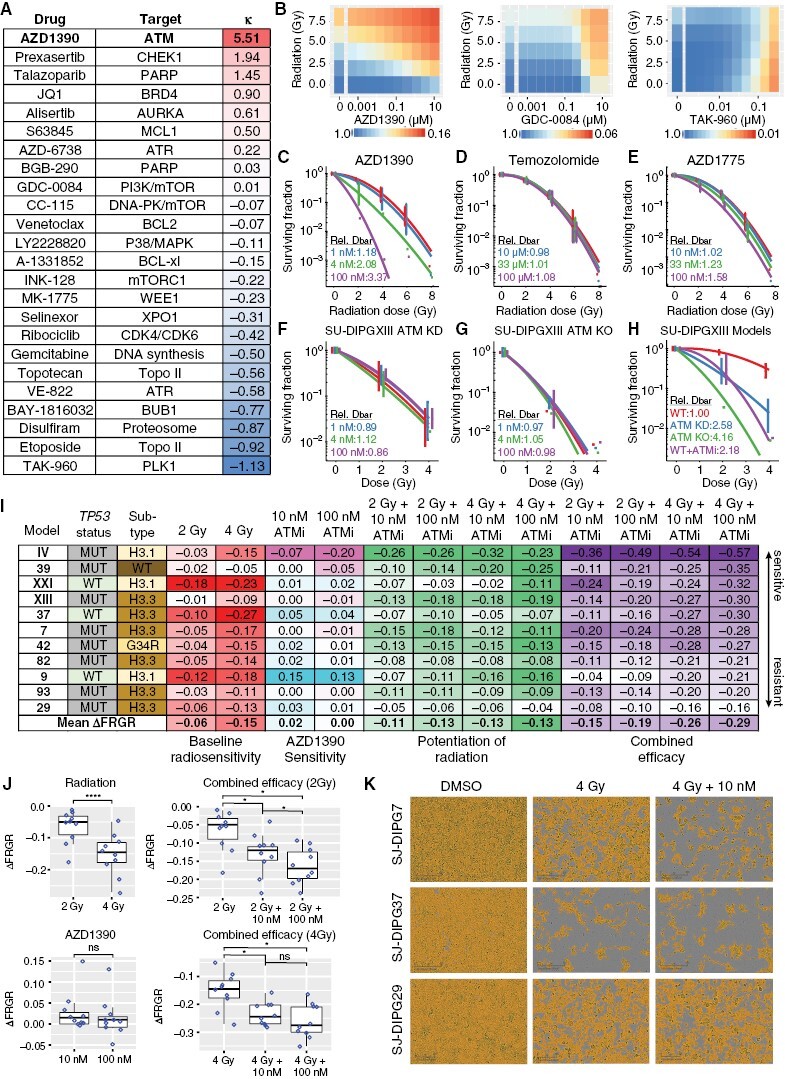
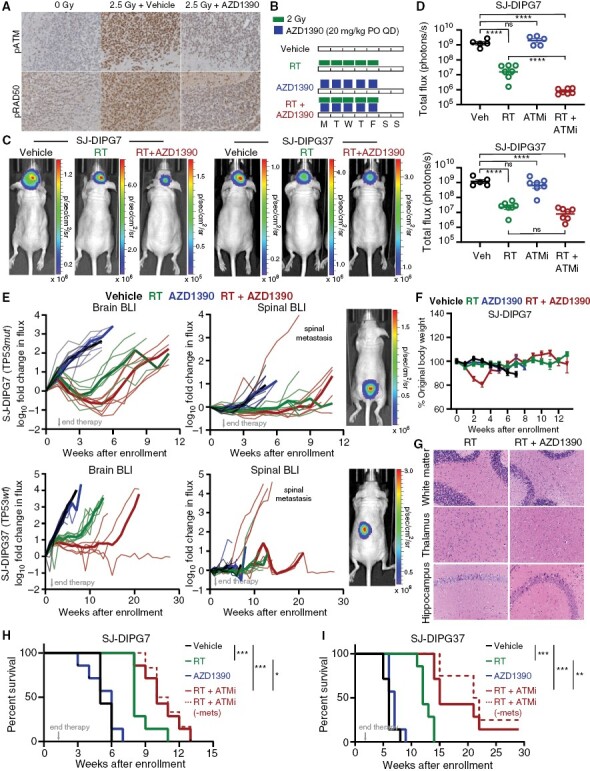
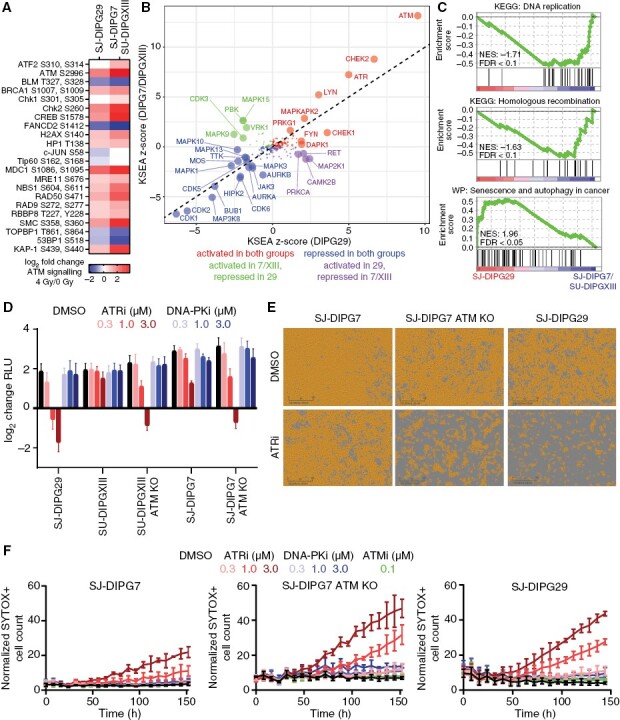
Methods: We conducted an unbiased screen in pHGG cells that combined radiation with clinical candidates targeting the DDR and identified the ATM inhibitor AZD1390. Subsequently, we profiled AZD1390 + radiation in an extensive panel of early passage pHGG cell lines, mechanistically characterized response to the combination in vitro in sensitive and resistant cells and evaluated the combination in vivo using TP53 wild-type and TP53 mutant orthotopic xenografts.
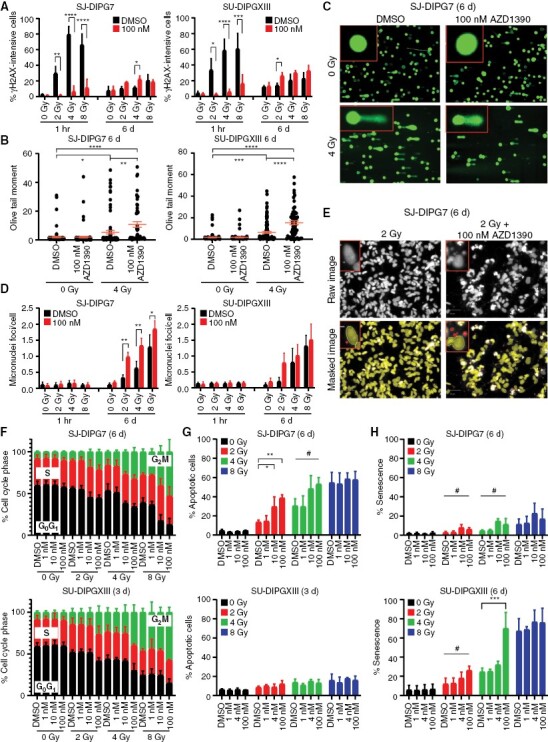
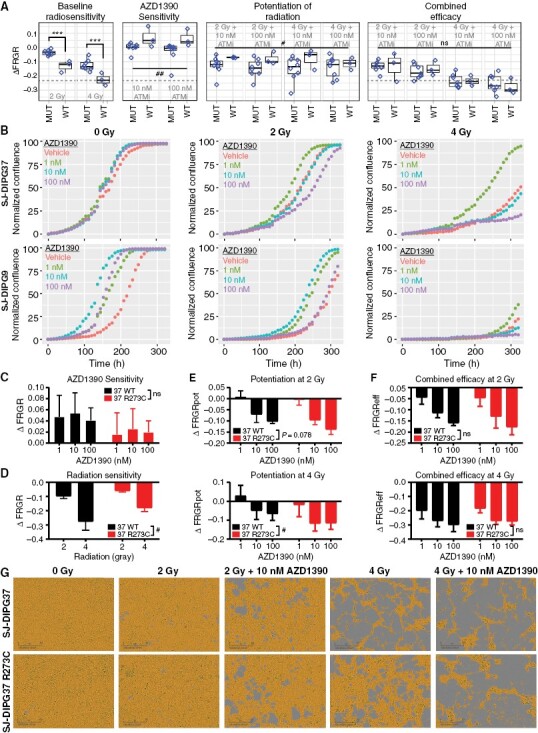
Results: AZD1390 significantly potentiated radiation across molecular subgroups of pHGG by increasing mutagenic nonhomologous end joining and augmenting genomic instability. In contrast to previous reports, ATM inhibition significantly improved the efficacy of radiation in both TP53 wild-type and TP53 mutant isogenic cell lines and distinct orthotopic xenograft models. Furthermore, we identified a novel mechanism of resistance to AZD1390 + radiation that was marked by an attenuated ATM pathway response which dampened sensitivity to ATM inhibition and induced synthetic lethality with ATR inhibition.
Conclusions: Our study supports the clinical evaluation of AZD1390 in combination with radiation in pediatric patients with HGG.
Keywords: ATM; DNA damage response; pediatric brain tumors; radiation therapy.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Society for Neuro-Oncology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Conflict of interest statement
Suzanne J. Baker owns stock in Astra Zeneca. The other authors declare no potential conflicts of interest.
Figures
References
-
- Juratli TA, Qin N, Cahill DP, Filbin MG.. Molecular pathogenesis and therapeutic implications in pediatric high-grade gliomas. Pharmacol Ther. 2018;182:70–79. - PubMed
-
- Mandell LR, Kadota R, Freeman C, et al. . There is no role for hyperfractionated radiotherapy in the management of children with newly diagnosed diffuse intrinsic brainstem tumors: results of a Pediatric Oncology Group phase III trial comparing conventional vs. hyperfractionated radiotherapy. Int J Radiat Oncol Biol Phys. 1999;43(5):959–964. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
