

Single cell analysis in head and neck cancer reveals potential immune evasion mechanisms during early metastasis
- PMID: 36973261
- PMCID: PMC10042873
- DOI: 10.1038/s41467-023-37379-y
Single cell analysis in head and neck cancer reveals potential immune evasion mechanisms during early metastasis
Abstract
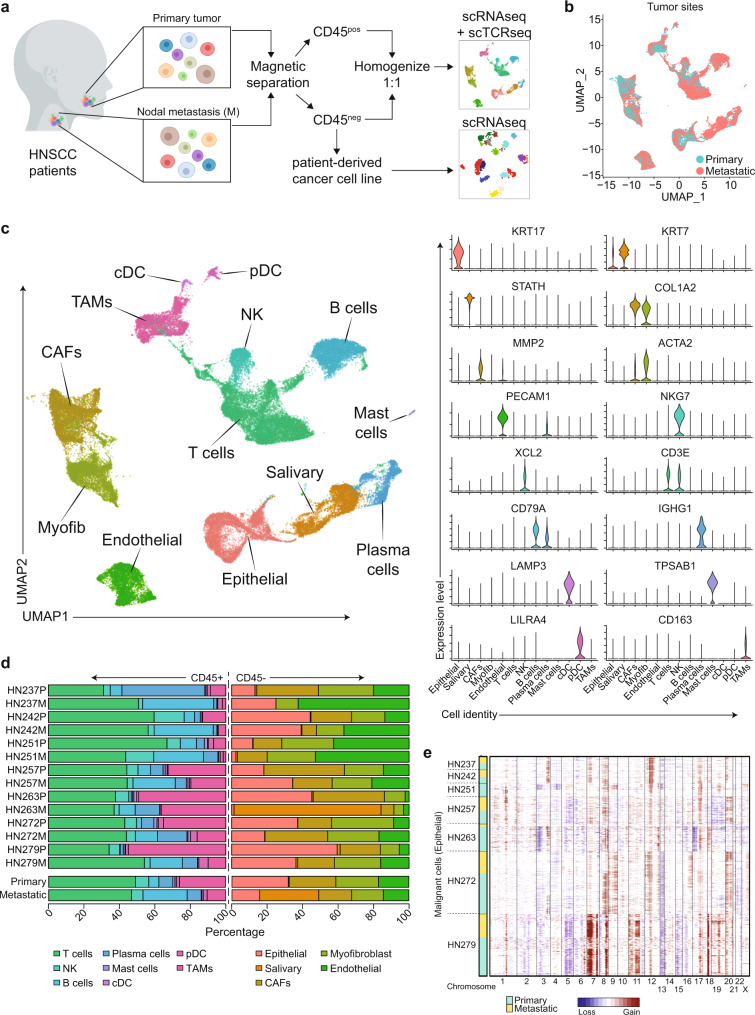
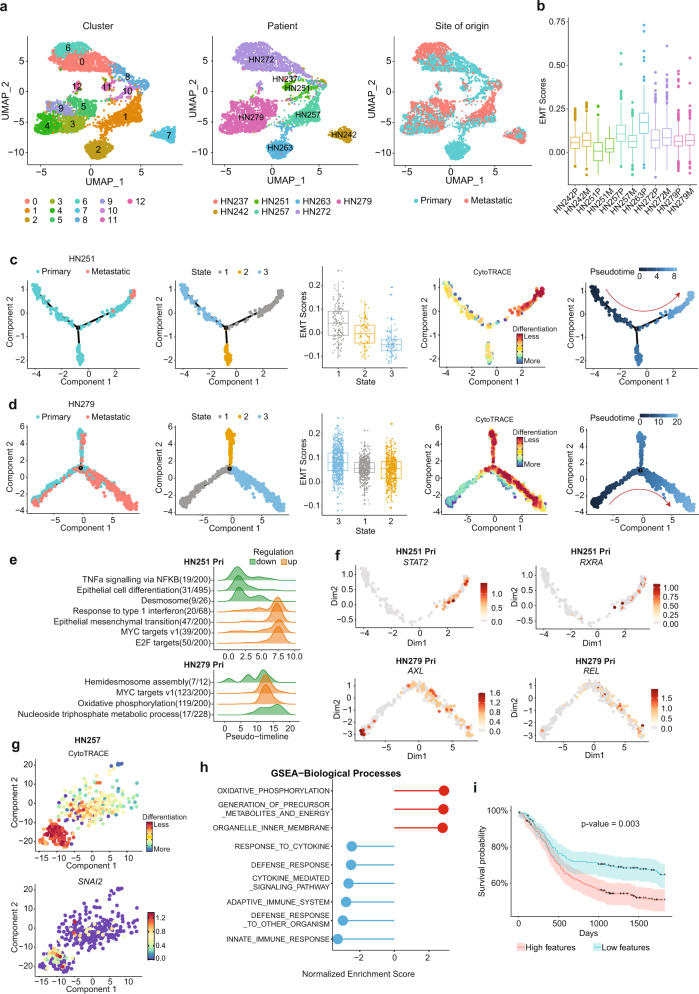
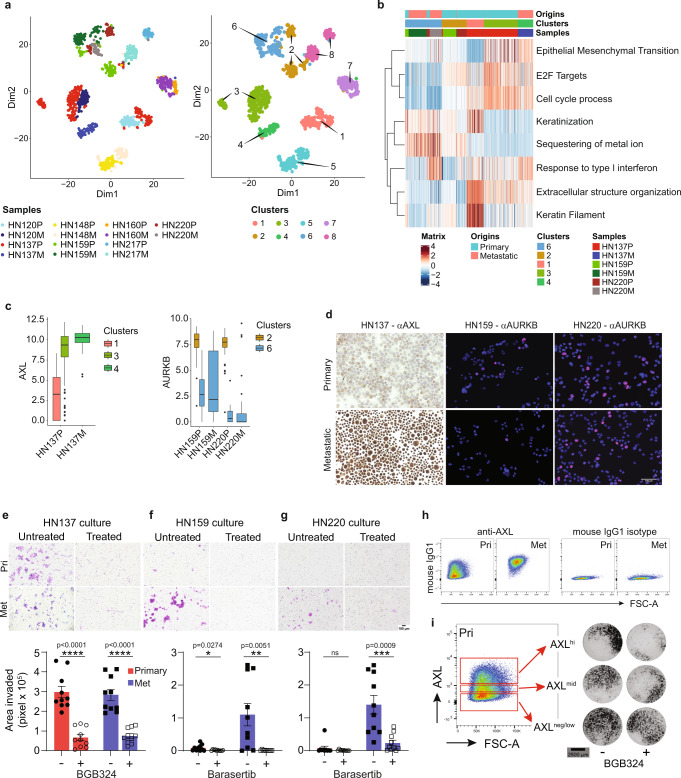
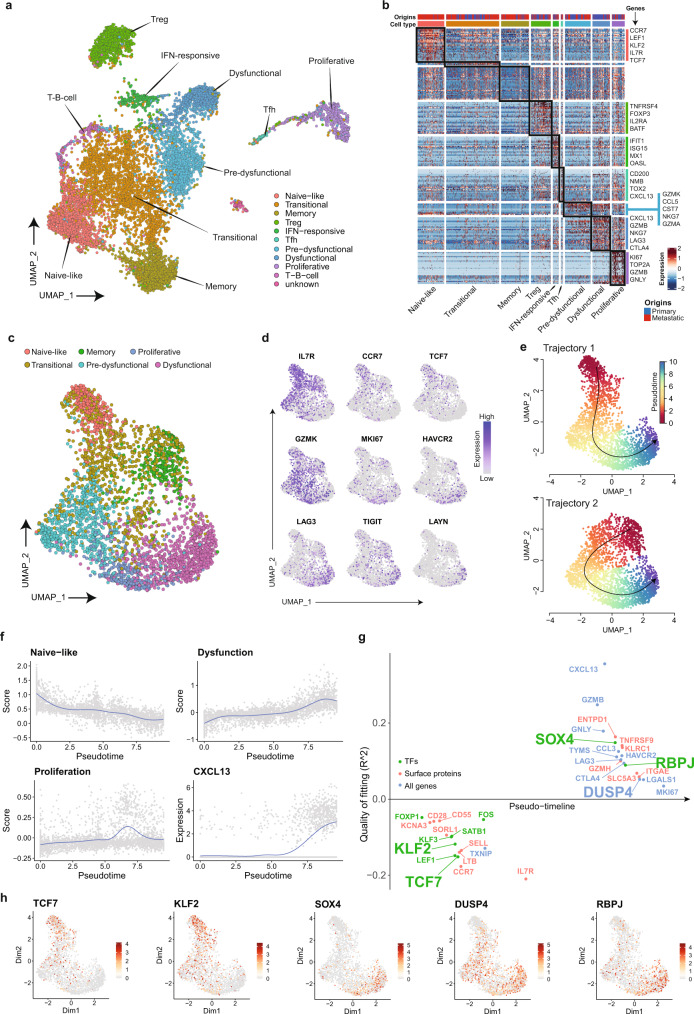
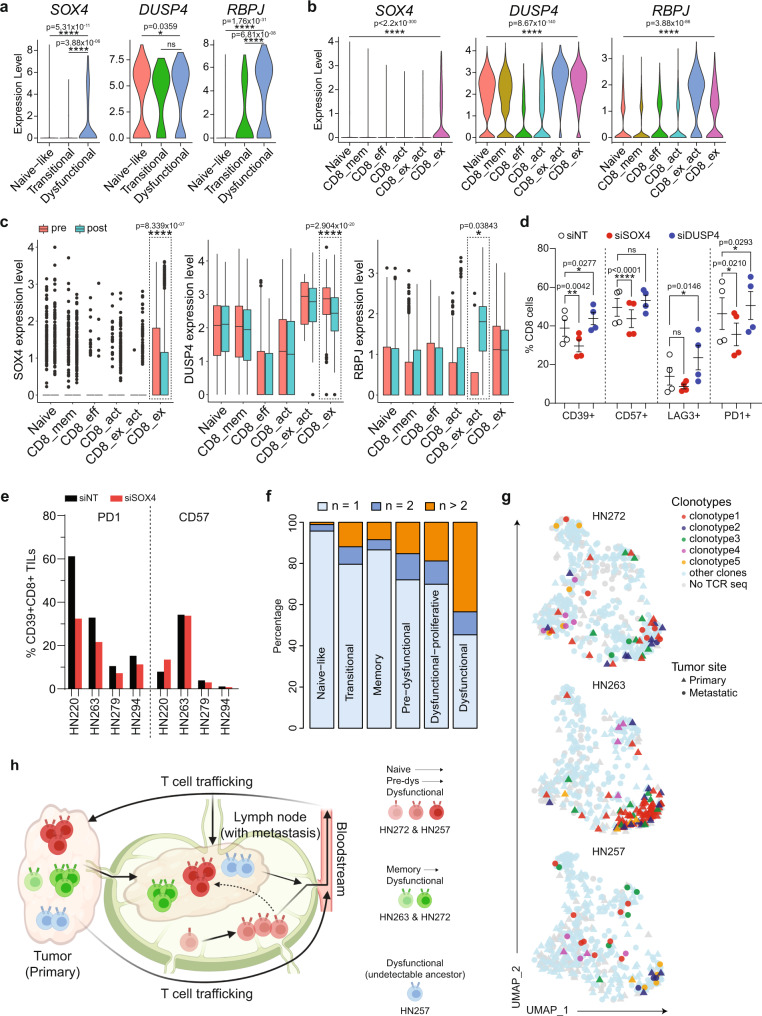
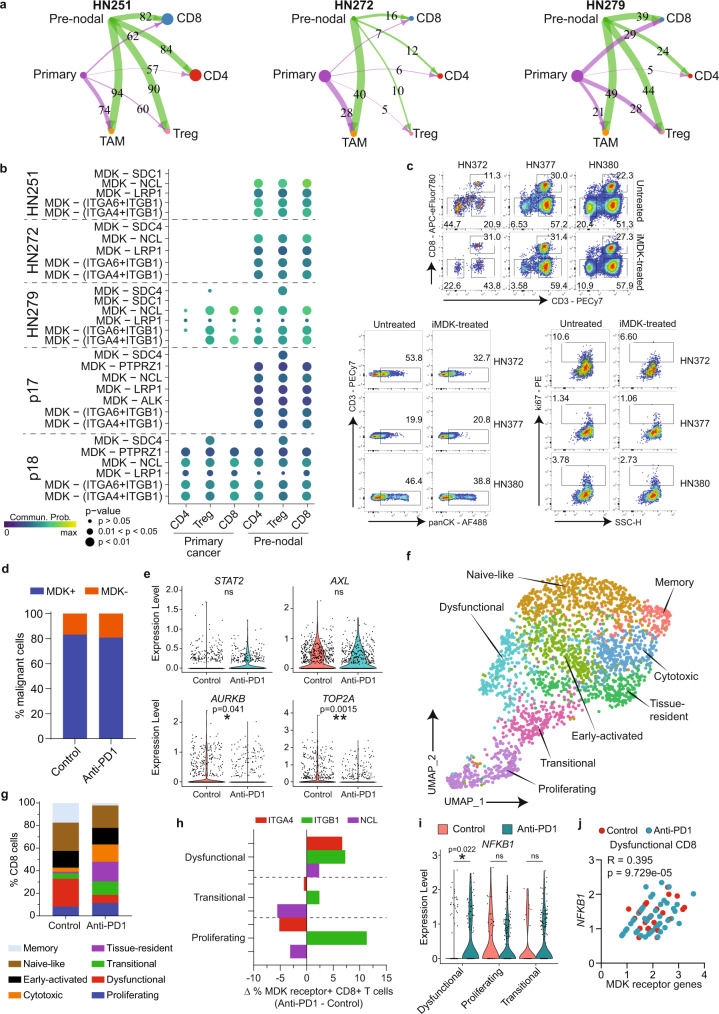
Profiling tumors at single-cell resolution provides an opportunity to understand complexities underpinning lymph-node metastases in head and neck squamous-cell carcinoma. Single-cell RNAseq (scRNAseq) analysis of cancer-cell trajectories identifies a subpopulation of pre-metastatic cells, driven by actionable pathways including AXL and AURK. Blocking these two proteins blunts tumor invasion in patient-derived cultures. Furthermore, scRNAseq analyses of tumor-infiltrating CD8 + T-lymphocytes show two distinct trajectories to T-cell dysfunction, corroborated by their clonal architecture based on single-cell T-cell receptor sequencing. By determining key modulators of these trajectories, followed by validation using external datasets and functional experiments, we uncover a role for SOX4 in mediating T-cell exhaustion. Finally, interactome analyses between pre-metastatic tumor cells and CD8 + T-lymphocytes uncover a putative role for the Midkine pathway in immune-modulation and this is confirmed by scRNAseq of tumors from humanized mice. Aside from specific findings, this study demonstrates the importance of tumor heterogeneity analyses in identifying key vulnerabilities during early metastasis.
© 2023. The Author(s).
Conflict of interest statement
NGI has/had a consulting or advisory role in PairX Therapeutics and Invitrocue PLC, and received honoraria from Kalbe Biotech and Agilent, all of which are outside this submitted work. DSWT received honoraria from Bristol-Myers Squibb, Takeda Pharmaceuticals, Novartis, Roche, and Pfizer; and has consulting or advisory role in Novartis, Merck, Loxo Oncology, AstraZeneca, Roche, and Pfizer. DSWT also received research funding from Novartis (Inst), GlaxoSmithKline (Inst), and AstraZeneca (Inst), outside this submitted work. None of the remaining authors have any other conflicts to report.
Figures
References
-
- Iyer NG, et al. Randomized trial comparing surgery and adjuvant radiotherapy versus concurrent chemoradiotherapy in patients with advanced, nonmetastatic squamous cell carcinoma of the head and neck: 10-year update and subset analysis. Cancer. 2015;121:1599–1607. doi: 10.1002/cncr.29251. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
