

Epigenetic and transcriptomic characterization reveals progression markers and essential pathways in clear cell renal cell carcinoma
- PMID: 36973268
- PMCID: PMC10042888
- DOI: 10.1038/s41467-023-37211-7
Epigenetic and transcriptomic characterization reveals progression markers and essential pathways in clear cell renal cell carcinoma
Erratum in
-
Author Correction: Epigenetic and transcriptomic characterization reveals progression markers and essential pathways in clear cell renal cell carcinoma.Nat Commun. 2023 May 17;14(1):2817. doi: 10.1038/s41467-023-38561-y. Nat Commun. 2023. PMID: 37198186 Free PMC article. No abstract available.
Abstract
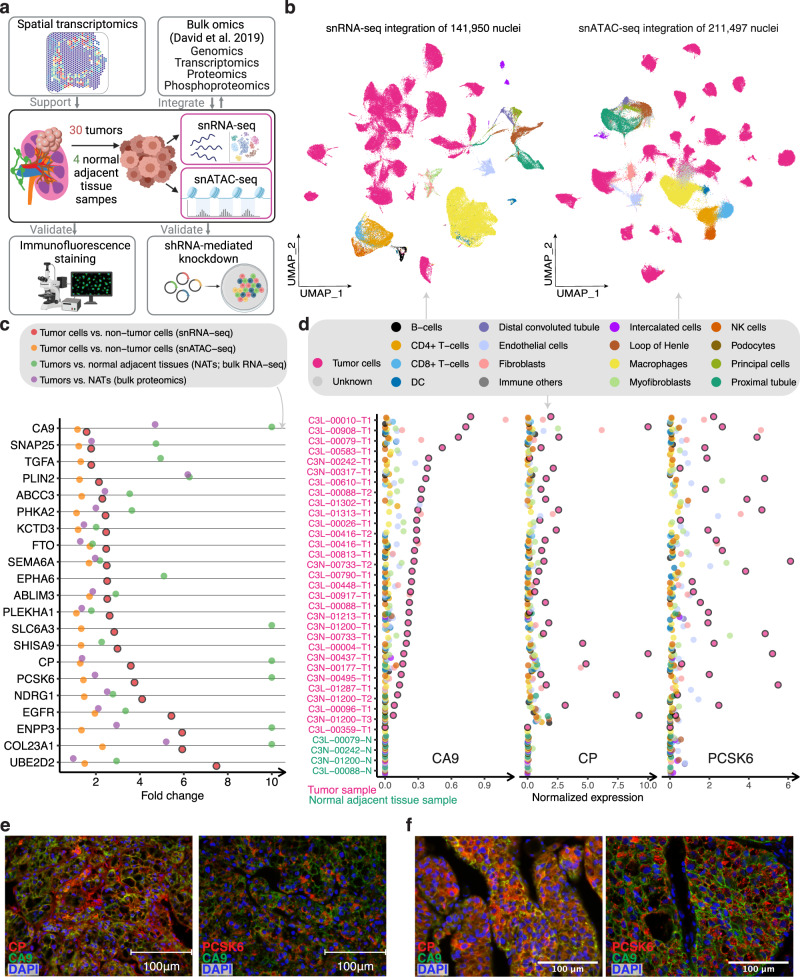
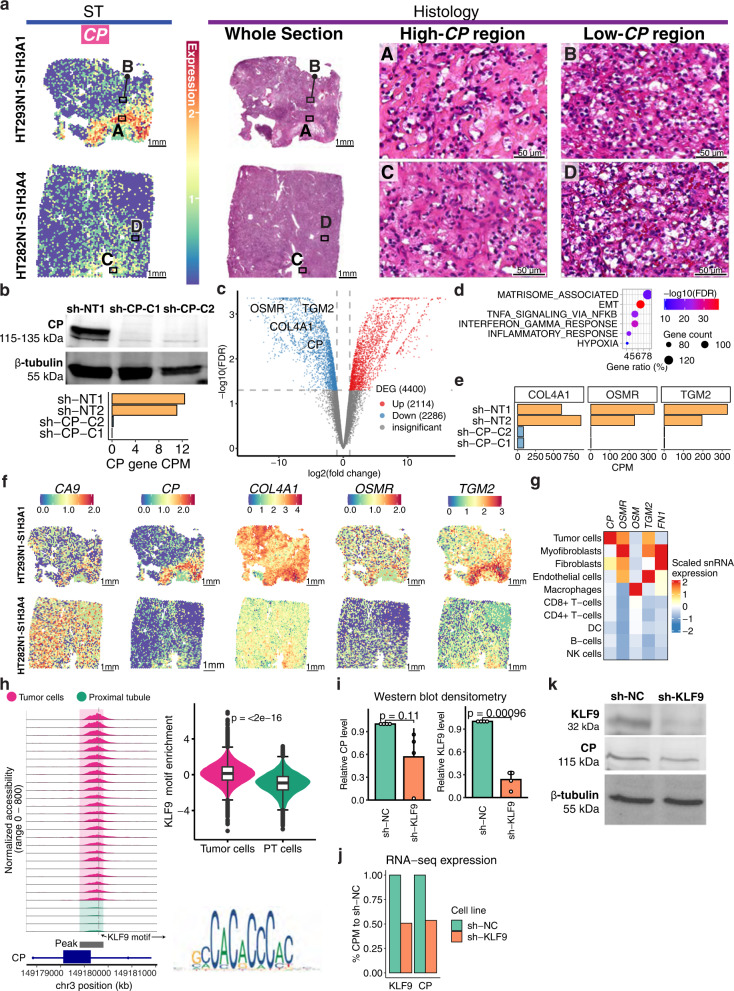
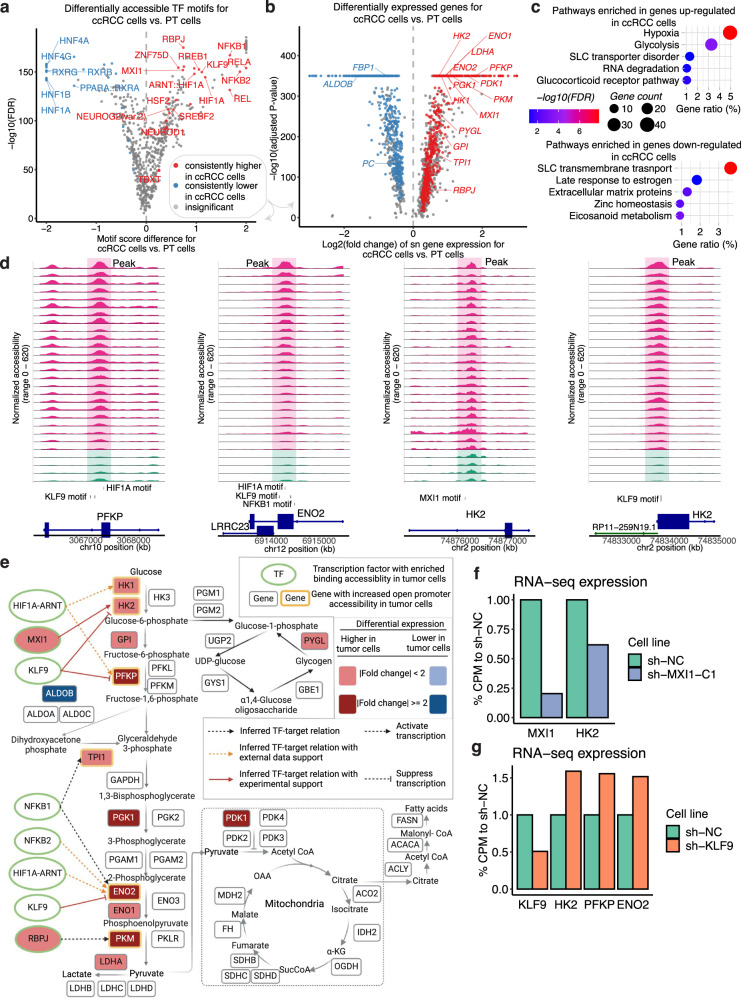
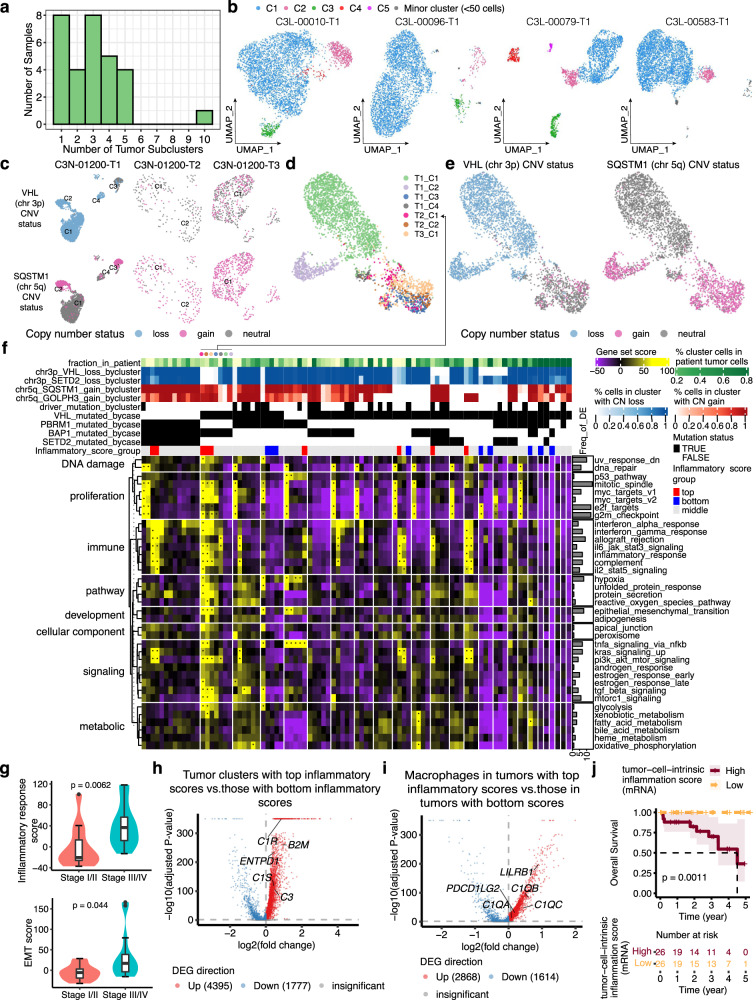
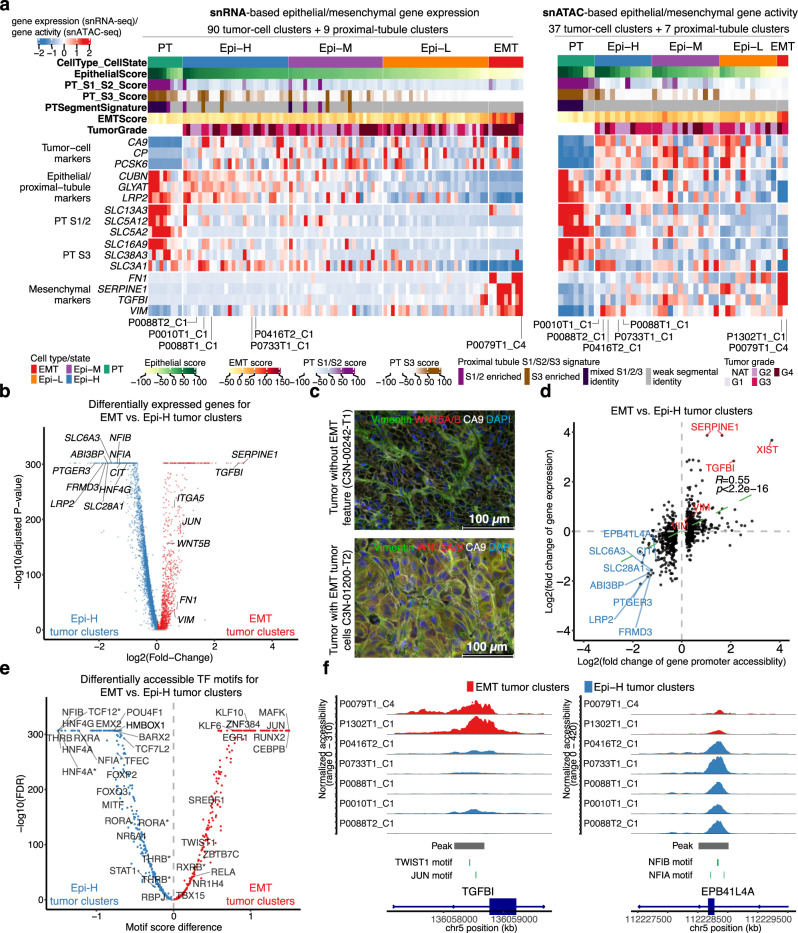
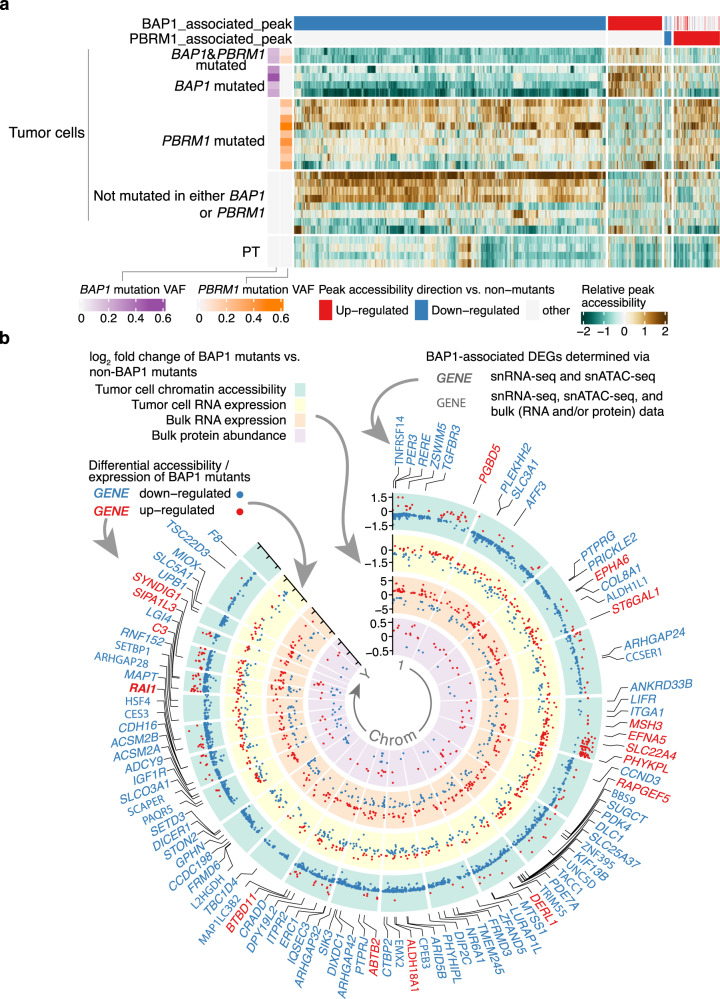
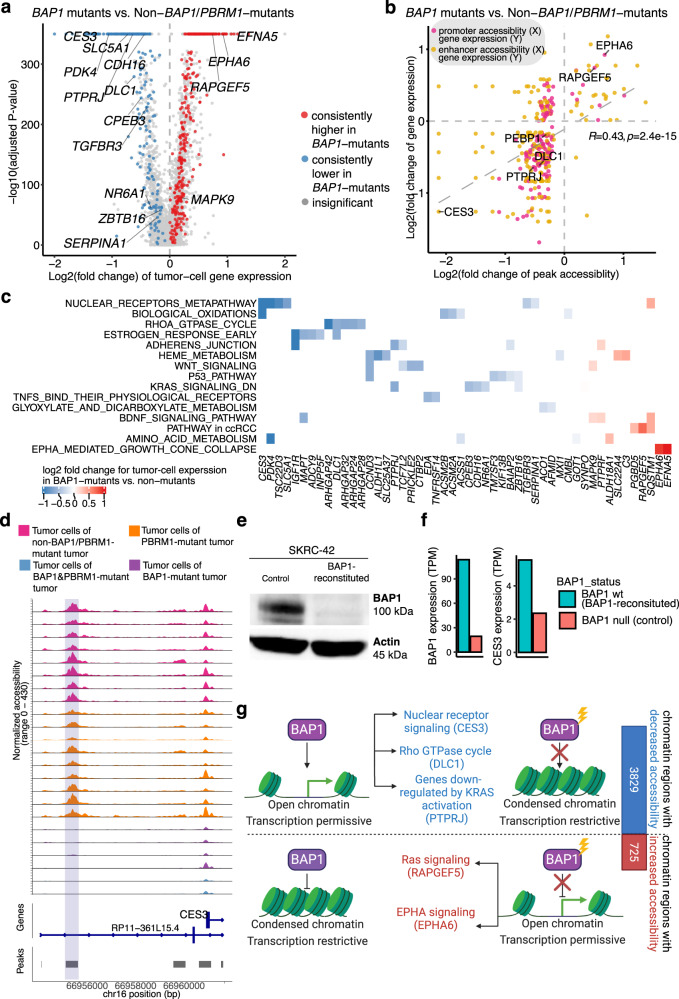
Identifying tumor-cell-specific markers and elucidating their epigenetic regulation and spatial heterogeneity provides mechanistic insights into cancer etiology. Here, we perform snRNA-seq and snATAC-seq in 34 and 28 human clear cell renal cell carcinoma (ccRCC) specimens, respectively, with matched bulk proteogenomics data. By identifying 20 tumor-specific markers through a multi-omics tiered approach, we reveal an association between higher ceruloplasmin (CP) expression and reduced survival. CP knockdown, combined with spatial transcriptomics, suggests a role for CP in regulating hyalinized stroma and tumor-stroma interactions in ccRCC. Intratumoral heterogeneity analysis portrays tumor cell-intrinsic inflammation and epithelial-mesenchymal transition (EMT) as two distinguishing features of tumor subpopulations. Finally, BAP1 mutations are associated with widespread reduction of chromatin accessibility, while PBRM1 mutations generally increase accessibility, with the former affecting five times more accessible peaks than the latter. These integrated analyses reveal the cellular architecture of ccRCC, providing insights into key markers and pathways in ccRCC tumorigenesis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Ljungberg B, et al. EAU guidelines on renal cell carcinoma: 2014 update. Eur. Urol. 2015;67:913–924. - PubMed
-
- Oosterwijk E, et al. Monoclonal antibody G 250 recognizes a determinant present in renal-cell carcinoma and absent from normal kidney. Int. J. Cancer. 1986;38:489–494. - PubMed
-
- Ye H, et al. CD70 is a promising CAR-T cell target in patients with advanced renal cell carcinoma. J. Clin. Oncol. 2022;40:384–384.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
