

A comprehensive platform for analyzing longitudinal multi-omics data
- PMID: 36973282
- PMCID: PMC10041512
- DOI: 10.1038/s41467-023-37432-w
A comprehensive platform for analyzing longitudinal multi-omics data
Abstract
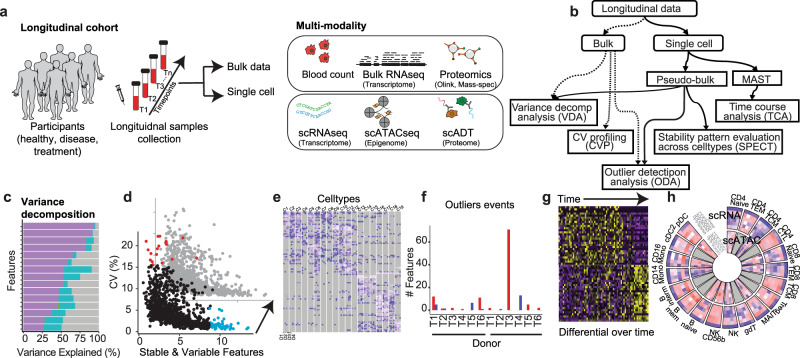
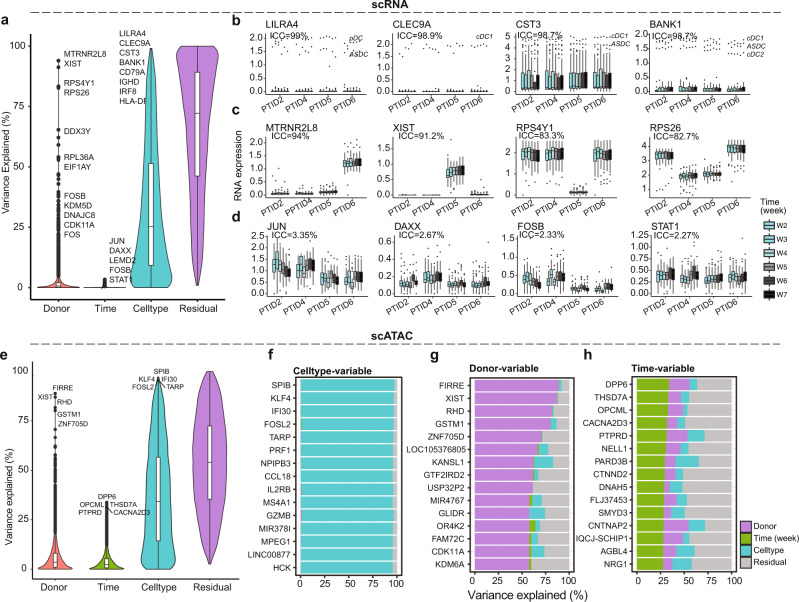
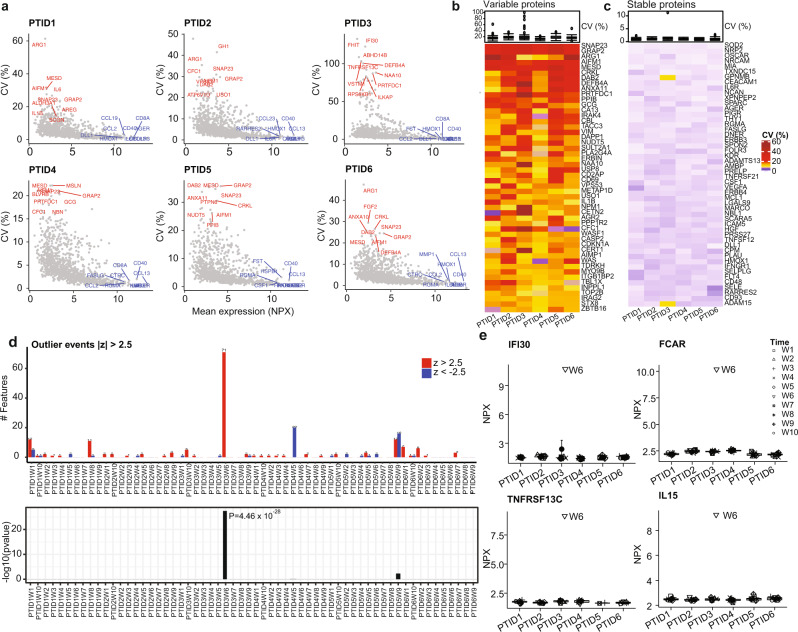
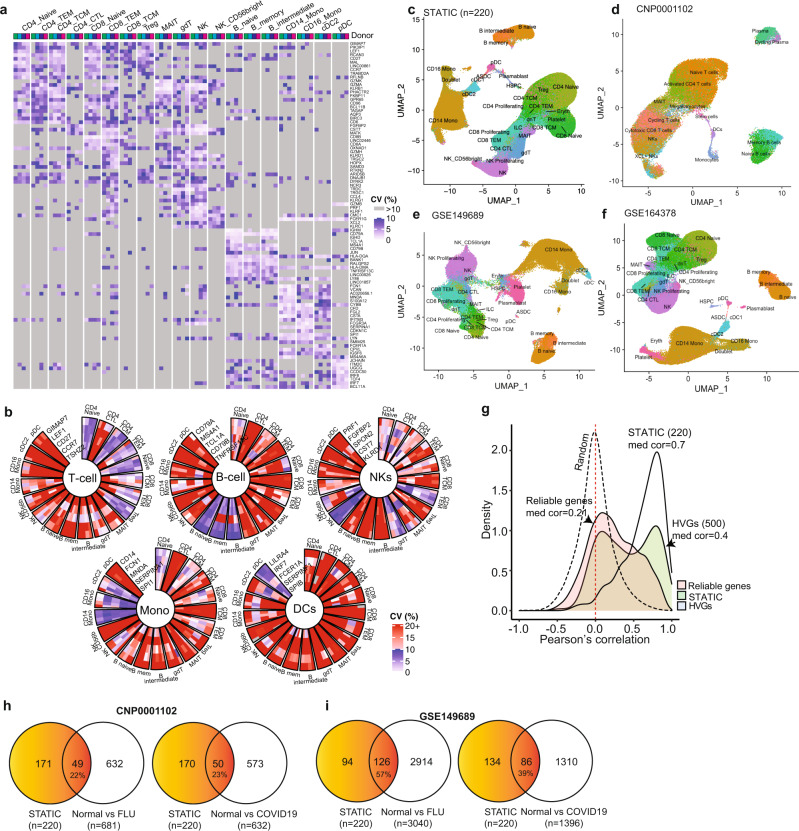
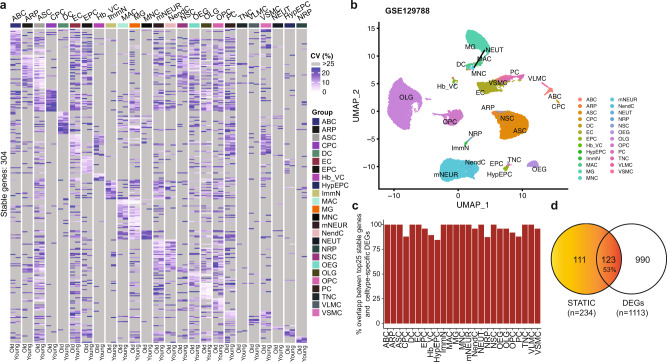
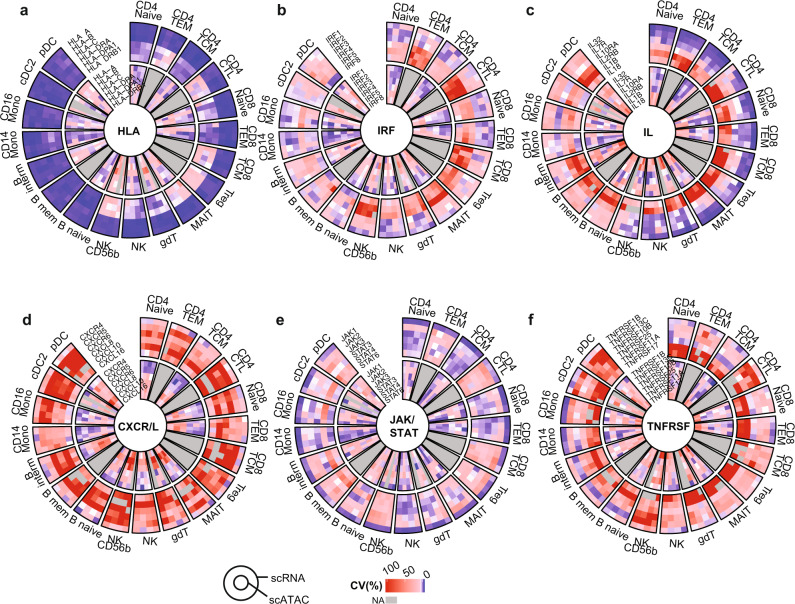
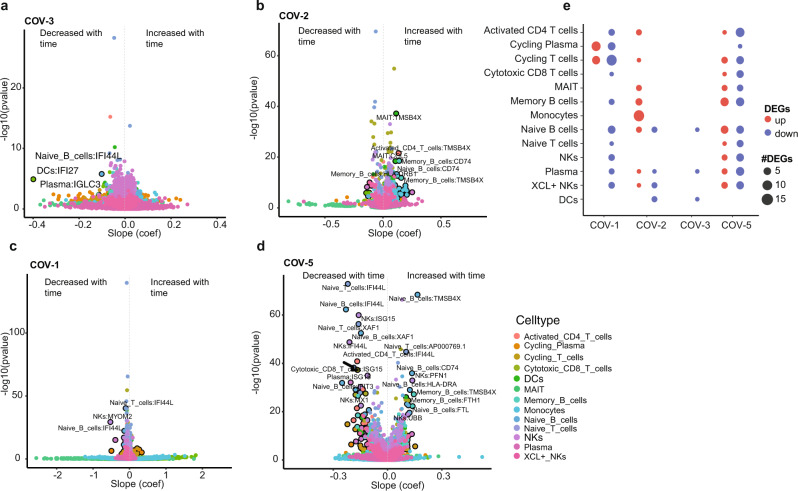
Longitudinal bulk and single-cell omics data is increasingly generated for biological and clinical research but is challenging to analyze due to its many intrinsic types of variations. We present PALMO ( https://github.com/aifimmunology/PALMO ), a platform that contains five analytical modules to examine longitudinal bulk and single-cell multi-omics data from multiple perspectives, including decomposition of sources of variations within the data, collection of stable or variable features across timepoints and participants, identification of up- or down-regulated markers across timepoints of individual participants, and investigation on samples of same participants for possible outlier events. We have tested PALMO performance on a complex longitudinal multi-omics dataset of five data modalities on the same samples and six external datasets of diverse background. Both PALMO and our longitudinal multi-omics dataset can be valuable resources to the scientific community.
© 2023. The Author(s).
Conflict of interest statement
S.V.V., A.S., T.T., P.S., T.F.B., and X.L. are listed as inventors in a US patent application “Molecular Signatures For Cell Typing And Monitoring Immune Health” (application No. 63/291,234) based on this work. C.L. is currently an employee of GlaxoSmithKline. The remaining authors declare no competing interests.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
