

Nucleotide metabolism: a pan-cancer metabolic dependency
- PMID: 36973407
- PMCID: PMC10041518
- DOI: 10.1038/s41568-023-00557-7
Nucleotide metabolism: a pan-cancer metabolic dependency
Abstract
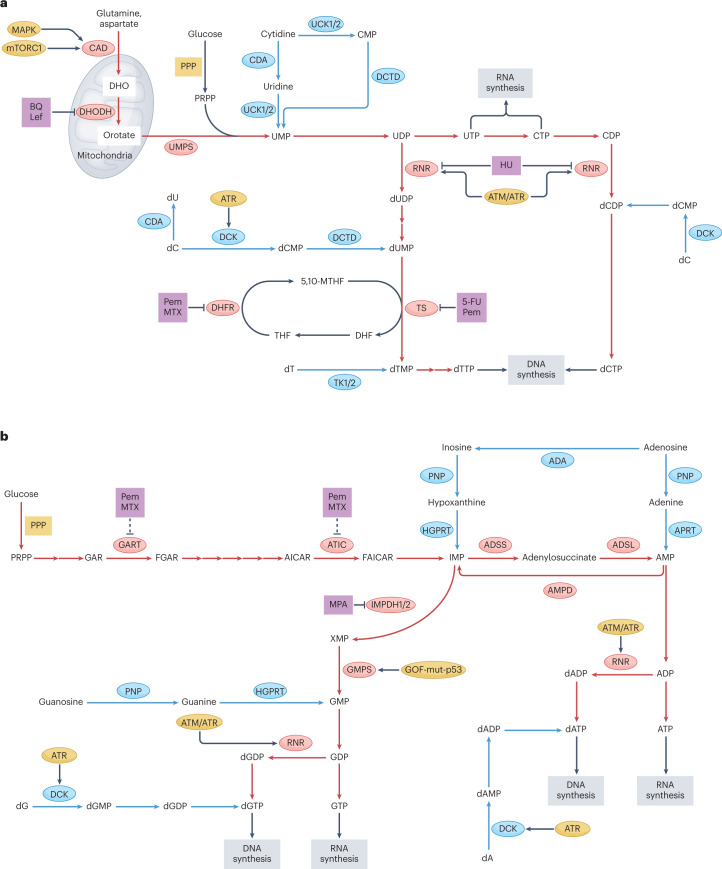
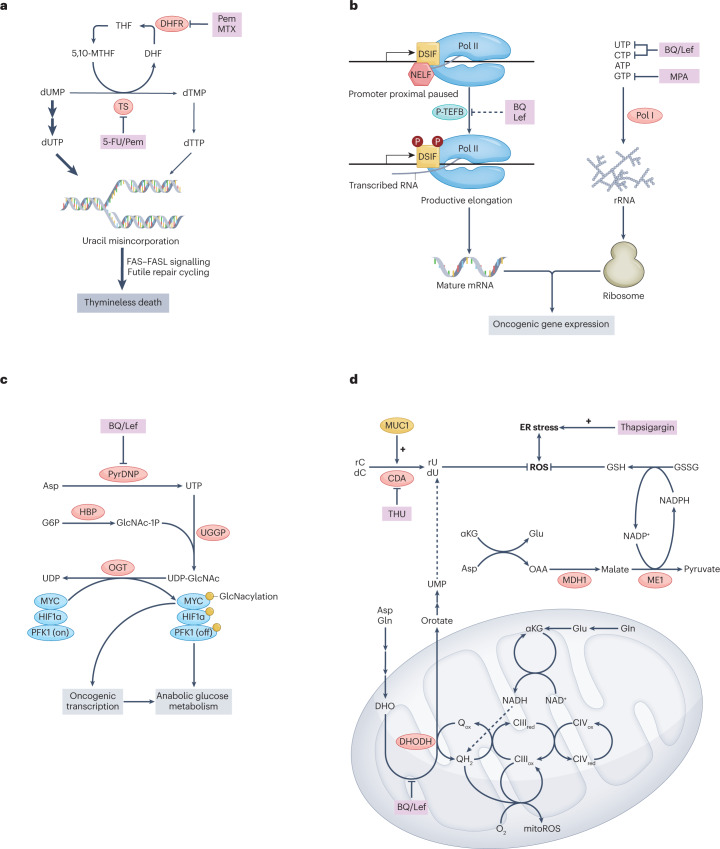
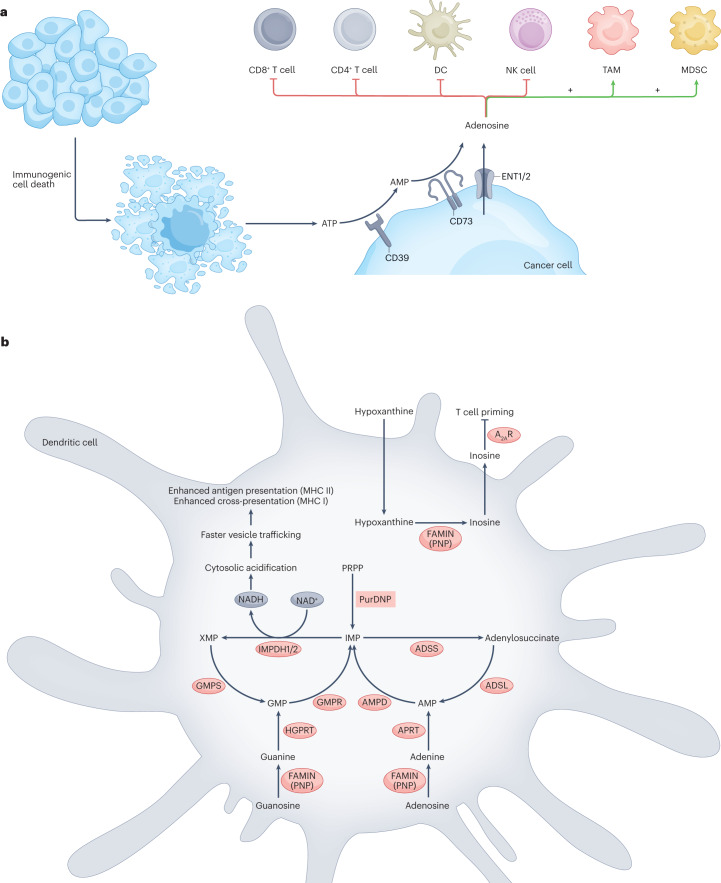
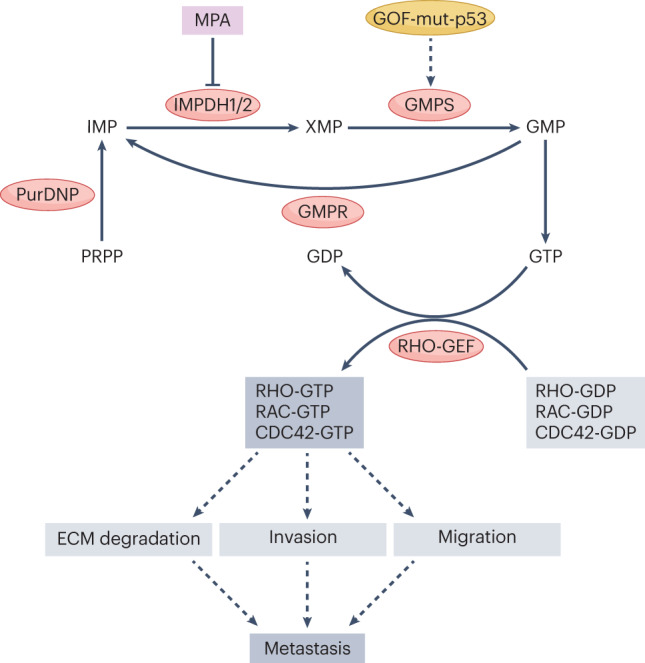
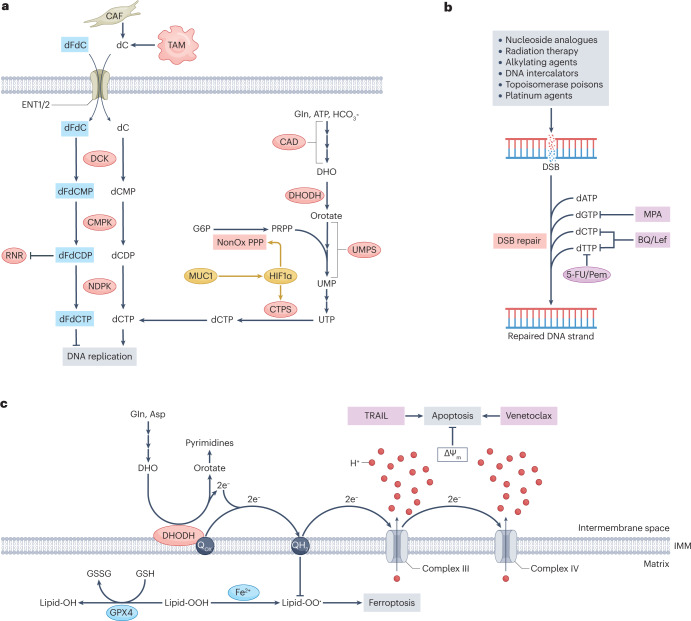
Metabolic alterations are a key hallmark of cancer cells, and the augmented synthesis and use of nucleotide triphosphates is a critical and universal metabolic dependency of cancer cells across different cancer types and genetic backgrounds. Many of the aggressive behaviours of cancer cells, including uncontrolled proliferation, chemotherapy resistance, immune evasion and metastasis, rely heavily on augmented nucleotide metabolism. Furthermore, most of the known oncogenic drivers upregulate nucleotide biosynthetic capacity, suggesting that this phenotype is a prerequisite for cancer initiation and progression. Despite the wealth of data demonstrating the efficacy of nucleotide synthesis inhibitors in preclinical cancer models and the well-established clinical use of these drugs in certain cancer settings, the full potential of these agents remains unrealized. In this Review, we discuss recent studies that have generated mechanistic insights into the diverse biological roles of hyperactive cancer cell nucleotide metabolism. We explore opportunities for combination therapies that are highlighted by these recent advances and detail key questions that remain to be answered, with the goal of informing urgently warranted future studies.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
