

Neuroblastoma arises in early fetal development and its evolutionary duration predicts outcome
- PMID: 36973454
- PMCID: PMC10101850
- DOI: 10.1038/s41588-023-01332-y
Neuroblastoma arises in early fetal development and its evolutionary duration predicts outcome
Abstract
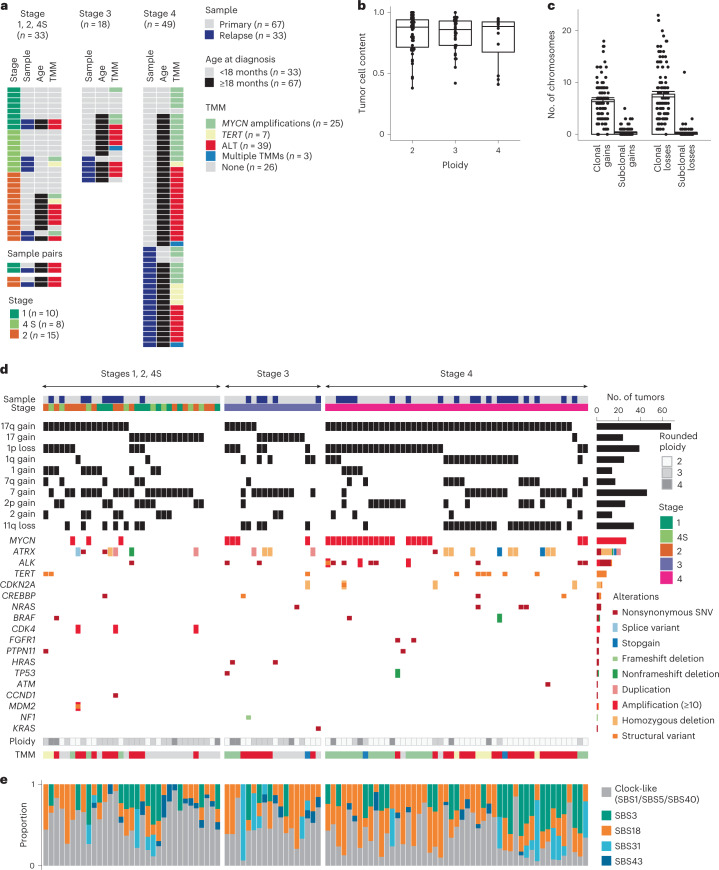
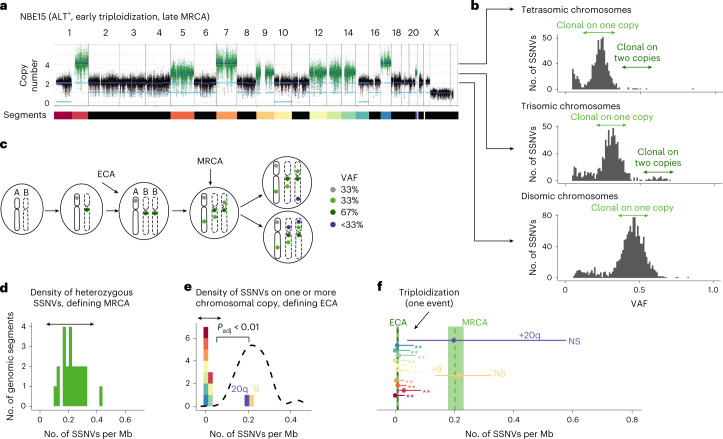
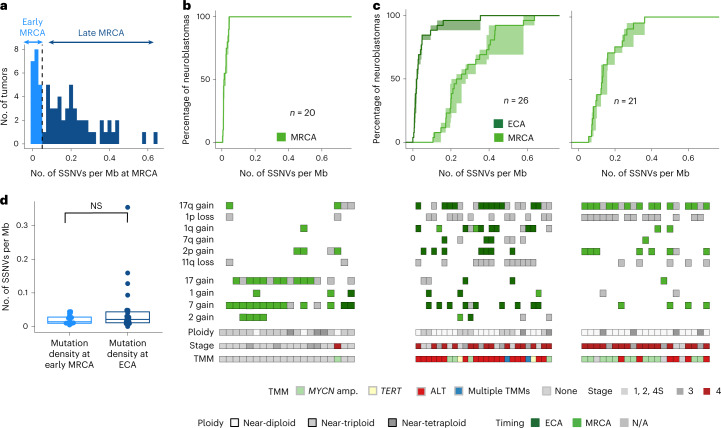
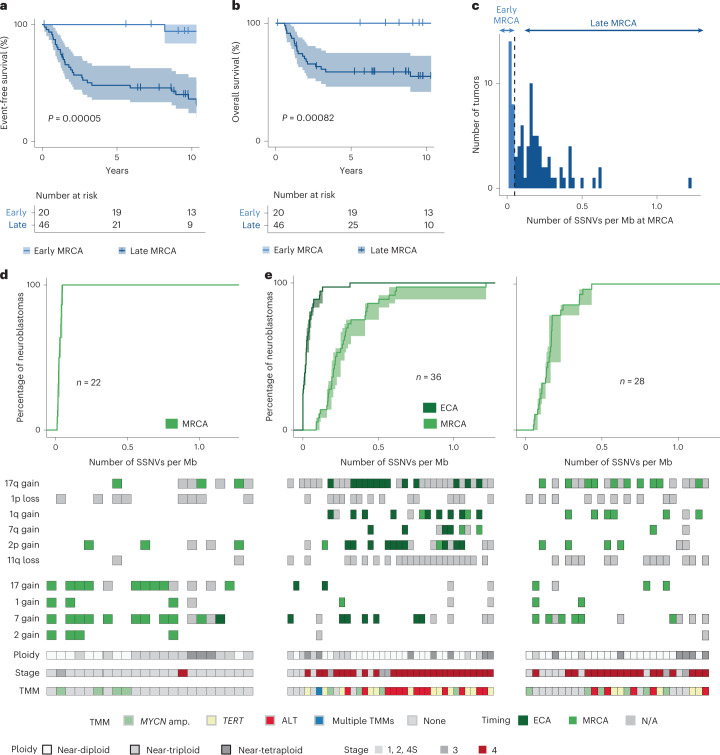
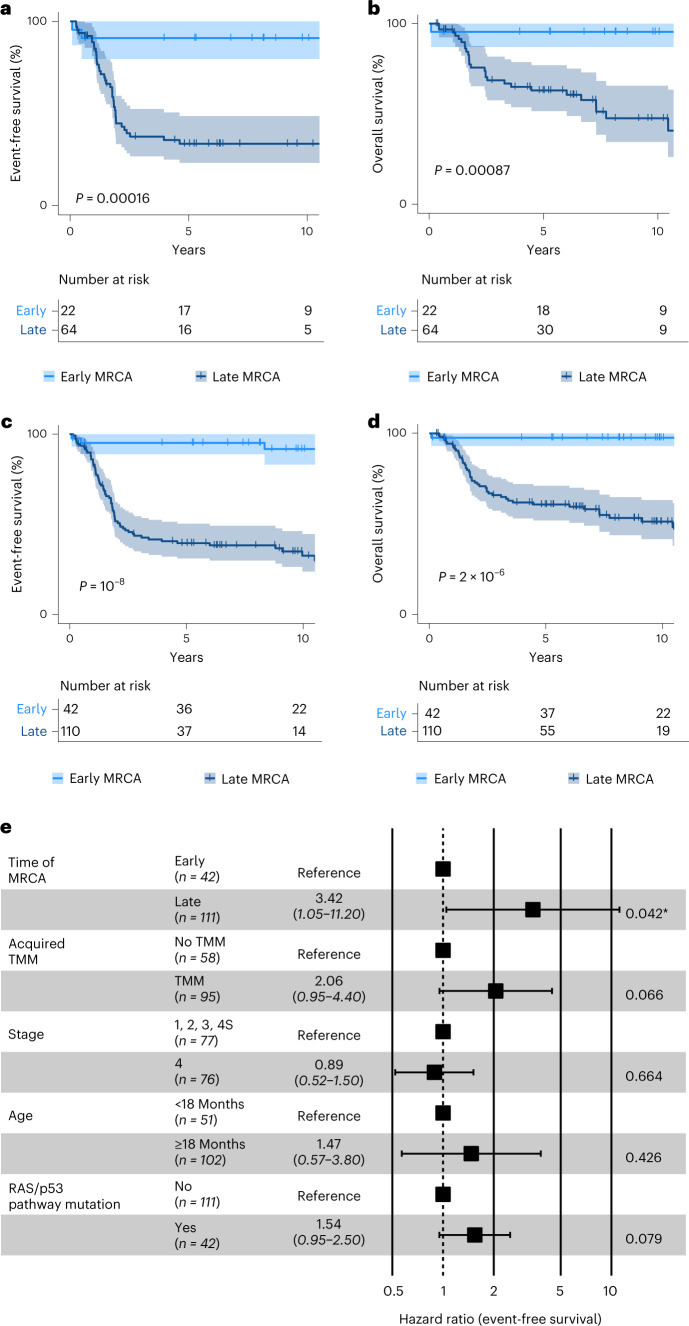
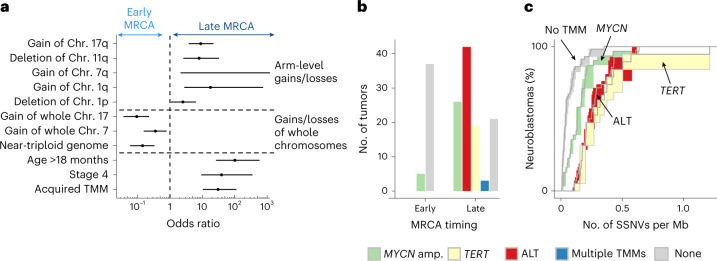
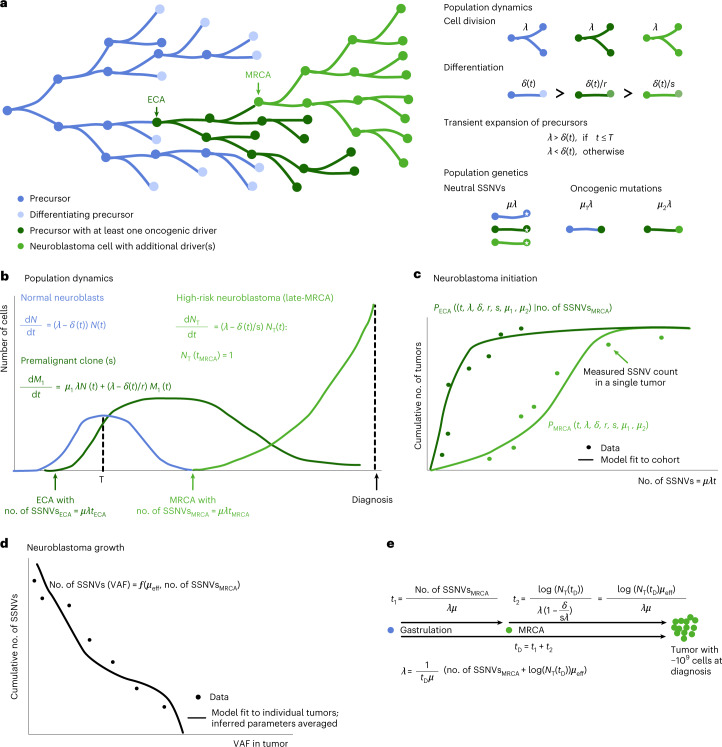
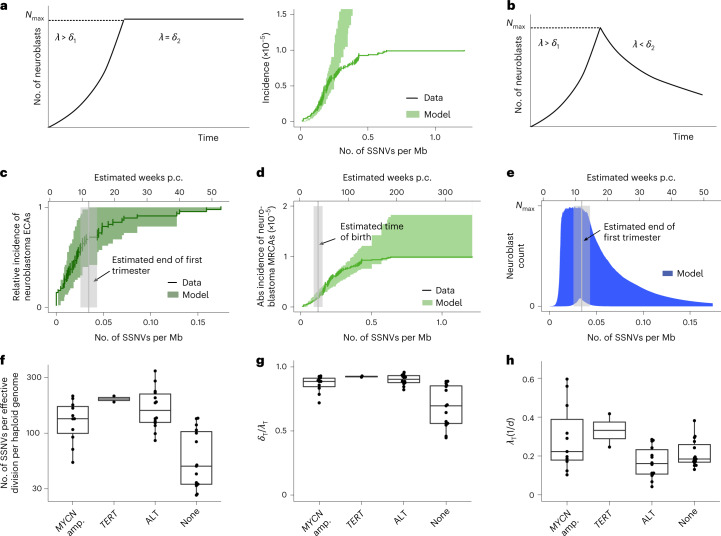
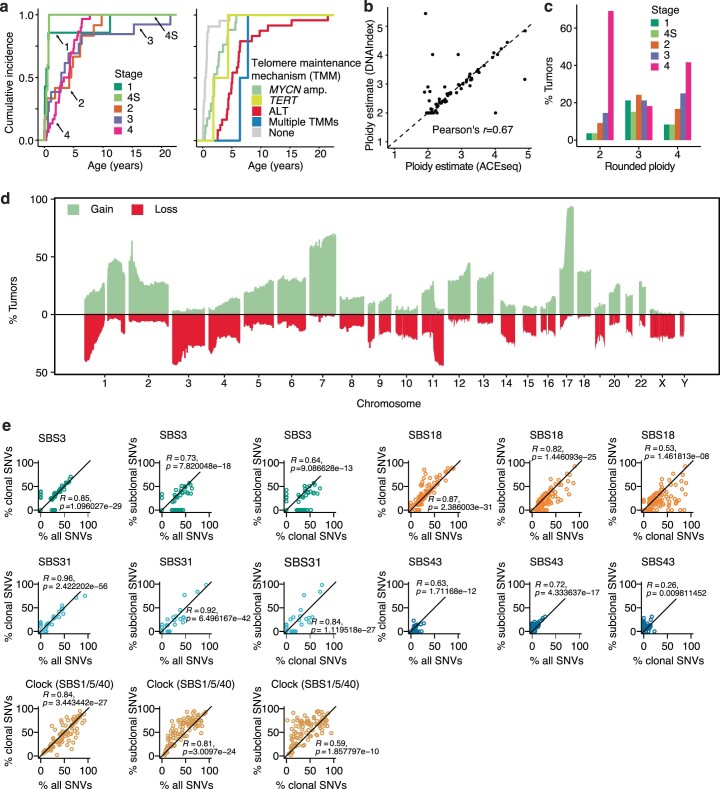
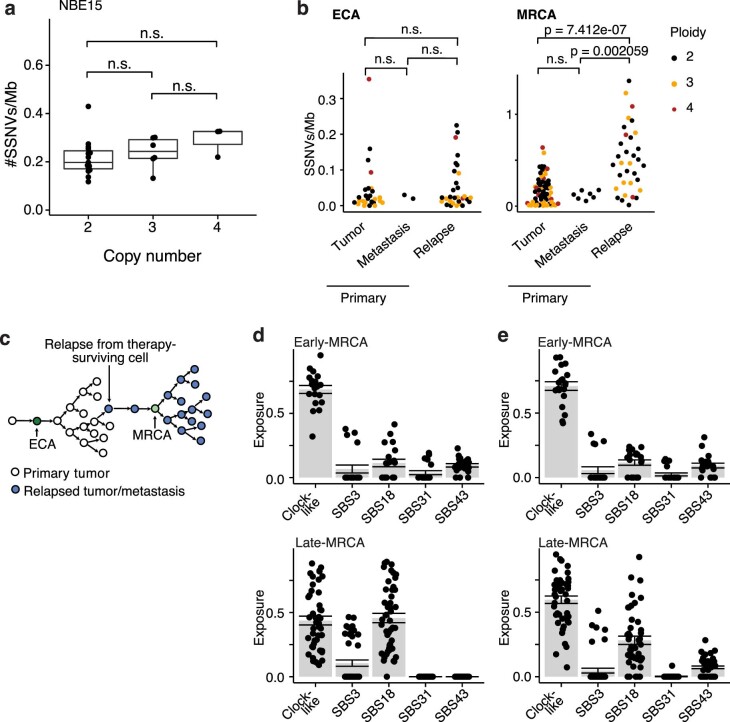
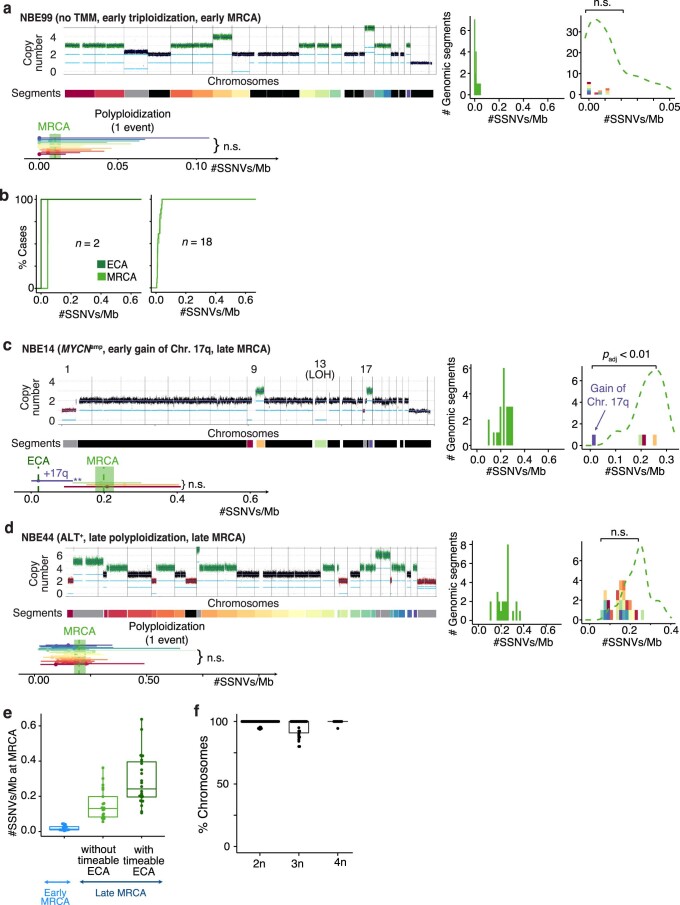
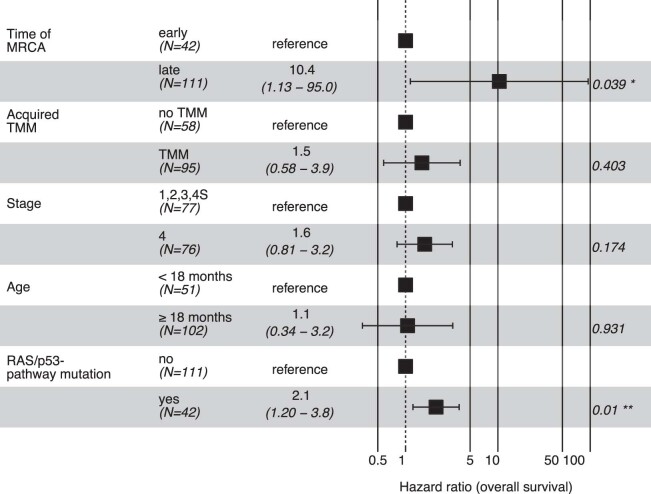
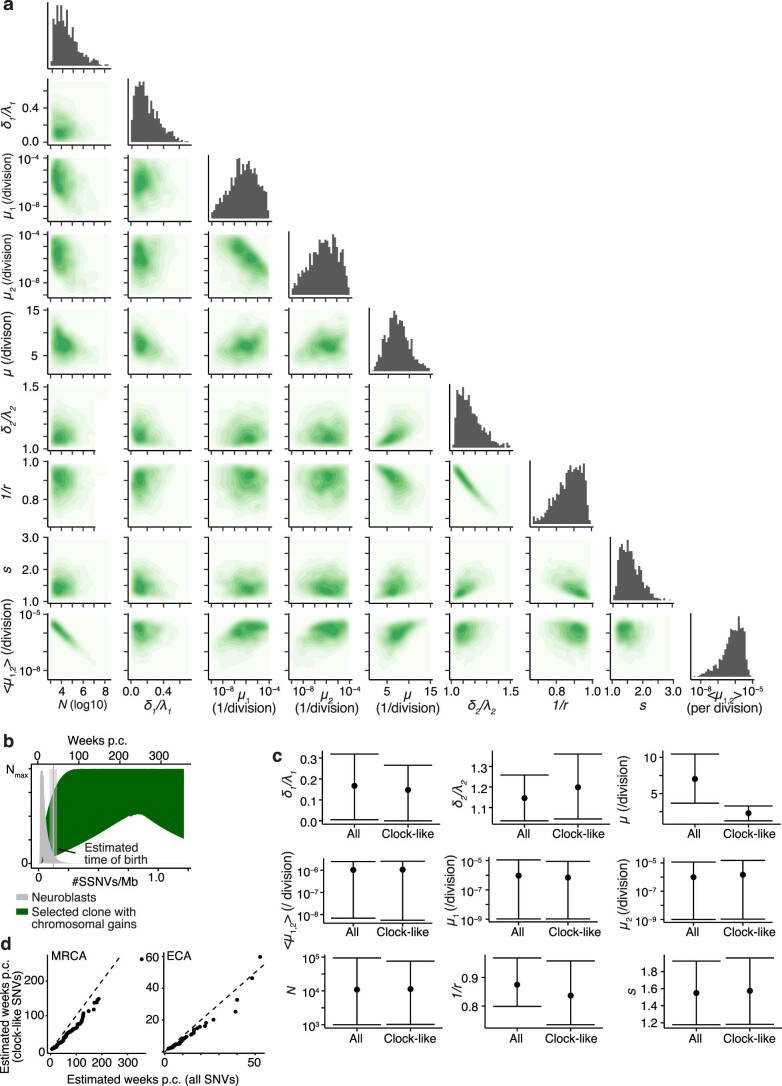
Neuroblastoma, the most frequent solid tumor in infants, shows very diverse outcomes from spontaneous regression to fatal disease. When these different tumors originate and how they evolve are not known. Here we quantify the somatic evolution of neuroblastoma by deep whole-genome sequencing, molecular clock analysis and population-genetic modeling in a comprehensive cohort covering all subtypes. We find that tumors across the entire clinical spectrum begin to develop via aberrant mitoses as early as the first trimester of pregnancy. Neuroblastomas with favorable prognosis expand clonally after short evolution, whereas aggressive neuroblastomas show prolonged evolution during which they acquire telomere maintenance mechanisms. The initial aneuploidization events condition subsequent evolution, with aggressive neuroblastoma exhibiting early genomic instability. We find in the discovery cohort (n = 100), and validate in an independent cohort (n = 86), that the duration of evolution is an accurate predictor of outcome. Thus, insight into neuroblastoma evolution may prospectively guide treatment decisions.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Mathematical modeling of neuroblastoma associates evolutionary patterns with outcomes.Nat Genet. 2023 Apr;55(4):530-531. doi: 10.1038/s41588-023-01358-2. Nat Genet. 2023. PMID: 36973453 No abstract available.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
