

Bridging the gaps: recent advances in diagnosis, care, and outcomes in congenital hyperinsulinism
- PMID: 36974442
- PMCID: PMC10330427
- DOI: 10.1097/MOP.0000000000001243
Bridging the gaps: recent advances in diagnosis, care, and outcomes in congenital hyperinsulinism
Abstract
Purpose of review: To highlight advances in congenital hyperinsulinism (HI), including newly described molecular mechanisms of disease, novel therapeutic interventions, and improved understanding of long-term outcomes.
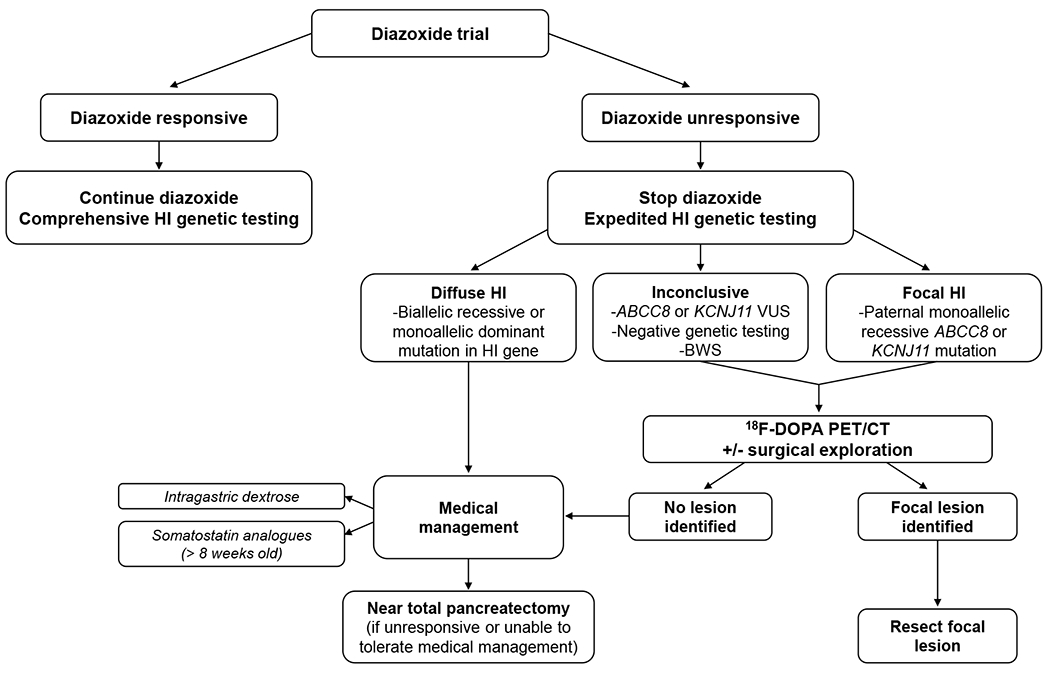
Recent findings: Important advances have been made elucidating the molecular mechanisms responsible for HI. Non-coding variants in HK1 have been found to cause aberrant hexokinase expression. Inactivating mutations in SLC25A36 have been identified in children with features of the hyperinsulinism hyperammonemia syndrome. Low-level mosaic mutations in known HI genes have been detected in cases of 'genetic testing negative' HI. Identification and localization of focal HI lesions remains a priority, since focal HI can be cured with surgery. Use of 68 Ga-NODAGA-exendin-4 PET has been proposed to localize focal lesions. Additional studies are needed before this technique replaces 18 F-DOPA PET as standard of care. Treatment options for children with diffuse HI remain limited. The long-acting somatostatin analog, lanreotide, was shown to significantly improve glycemic control in a large series of children with HI. New therapies are under development, with promising preliminary results. Long-term quality of life and neurodevelopmental outcomes remain suboptimal.
Summary: Advanced genetic and epigenomic analytic techniques have uncovered novel molecular mechanisms of HI. Development of new drugs holds promise to improve long-term outcomes for individuals with HI.
Copyright © 2023 Wolters Kluwer Health, Inc. All rights reserved.
Conflict of interest statement
Diva D. De Leon has served as principal investigator for industry-sponsored trials from Zealand Pharma, Hanmi Pharmaceuticals and Rezolute. She has received research funding from Twist Bioscience and Crinetics Pharmaceuticals. Diva D. De Leon has received consulting fees from Zealand Pharma, Crinetics Pharmaceuticals, Hanmi Pharmaceutical and Eiger Biopharmaceuticals. Diva D. De Leon is a named inventor in patents # USA Patent Number 9,616,108, 2017, USA Patent Number 9,821,031, 2017, Europe Patent Number EP 2120994, 2018, and Europe Patent Number EP2818181, 2019. These funders played no role in the completion of this manuscript.
Figures

References
-
- Stanley CA, Baker L. Hyperinsulinism in infancy: diagnosis by demonstration of abnormal response to fasting hypoglycemia. Pediatrics. 1976;57(5):702–11. - PubMed
-
- Quarrie Mc. Idiopathic spontaneously occurring hypoglycemia in infants; clinical significance of problem and treatment. AMA Am J Dis Child. 1954;87(4):399–428. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
