

SARS-CoV-2 variant identification using a genome tiling array and genotyping probes
- PMID: 36974726
- PMCID: PMC10350312
- DOI: 10.2217/pme-2022-0013
SARS-CoV-2 variant identification using a genome tiling array and genotyping probes
Abstract
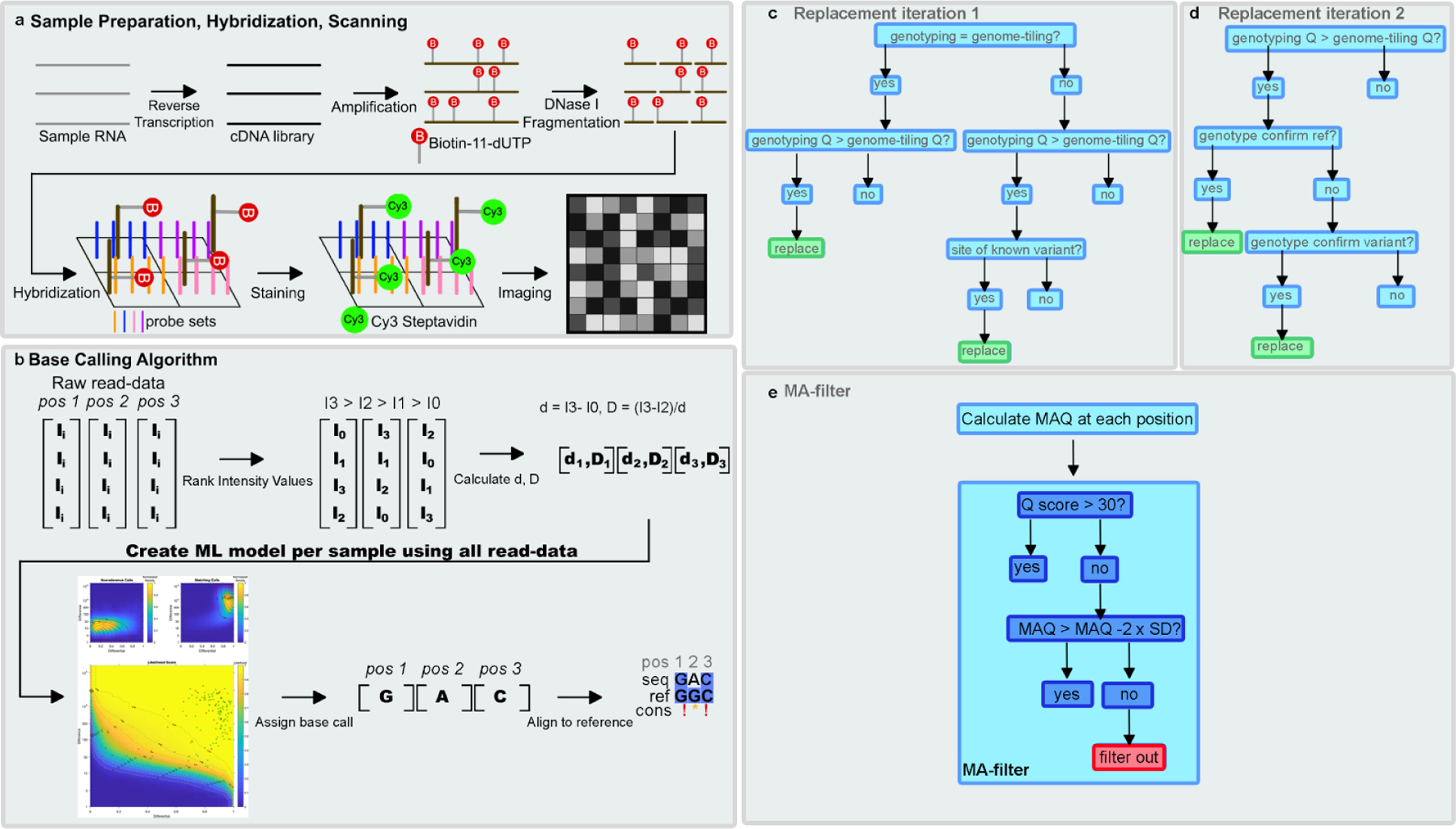
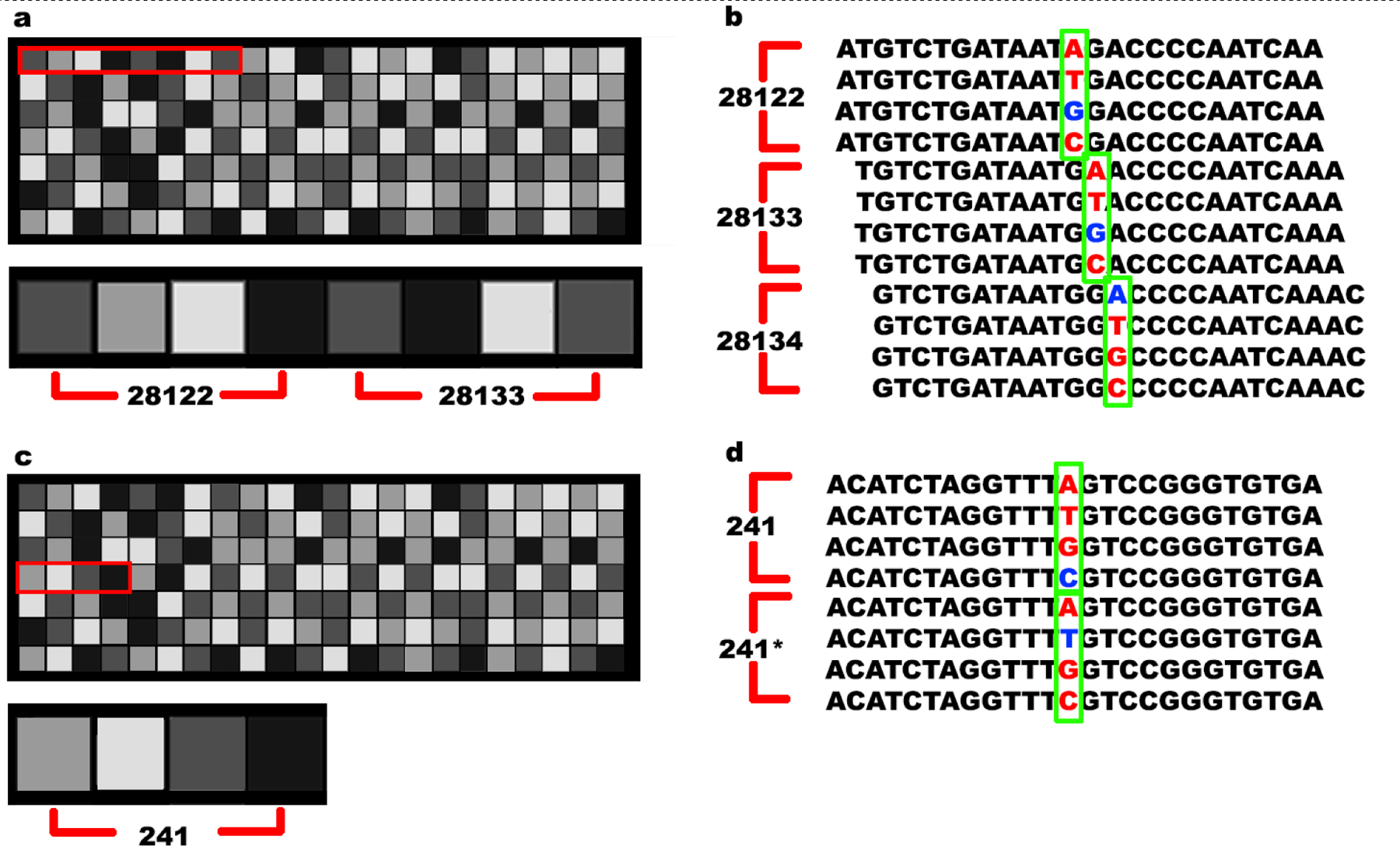
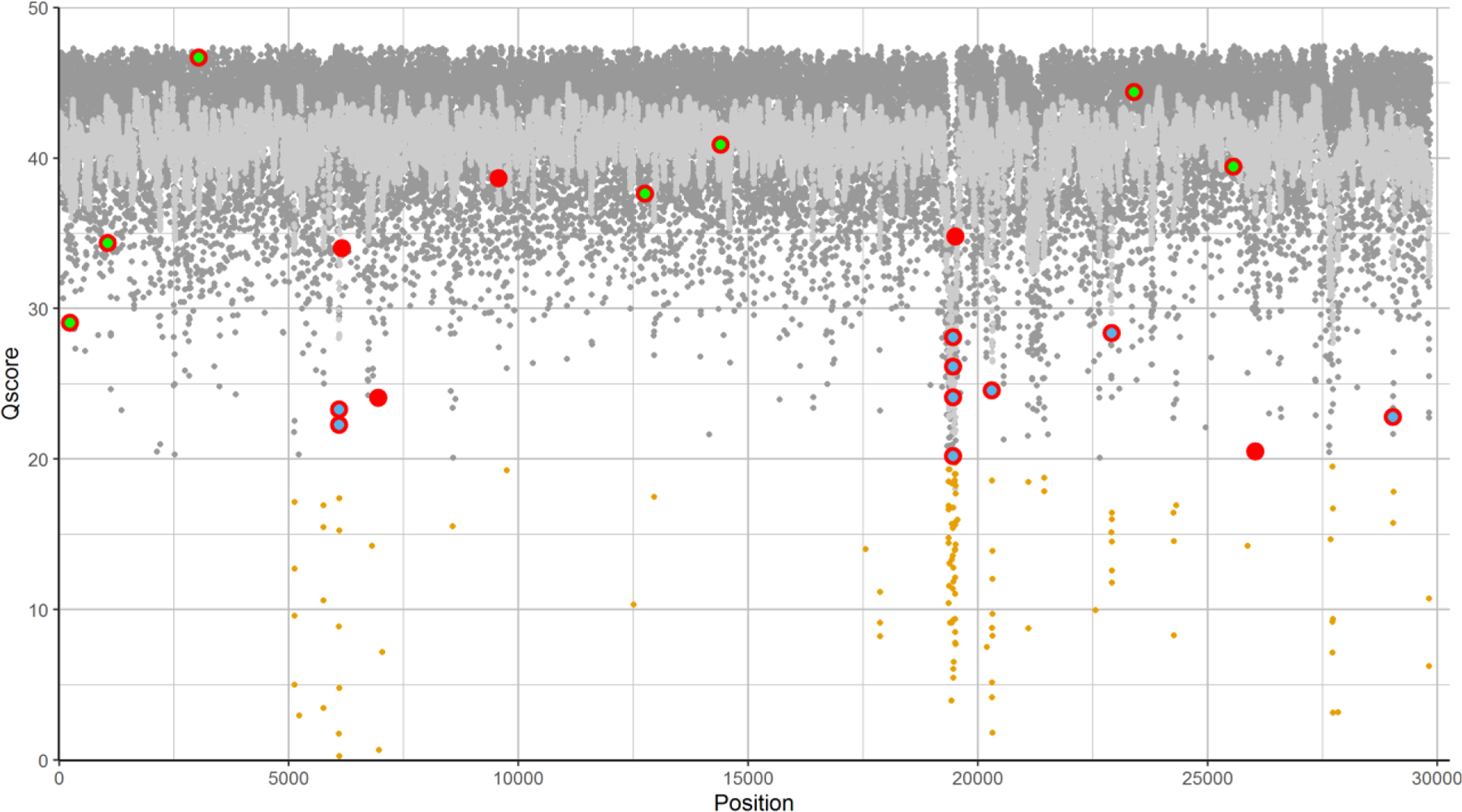
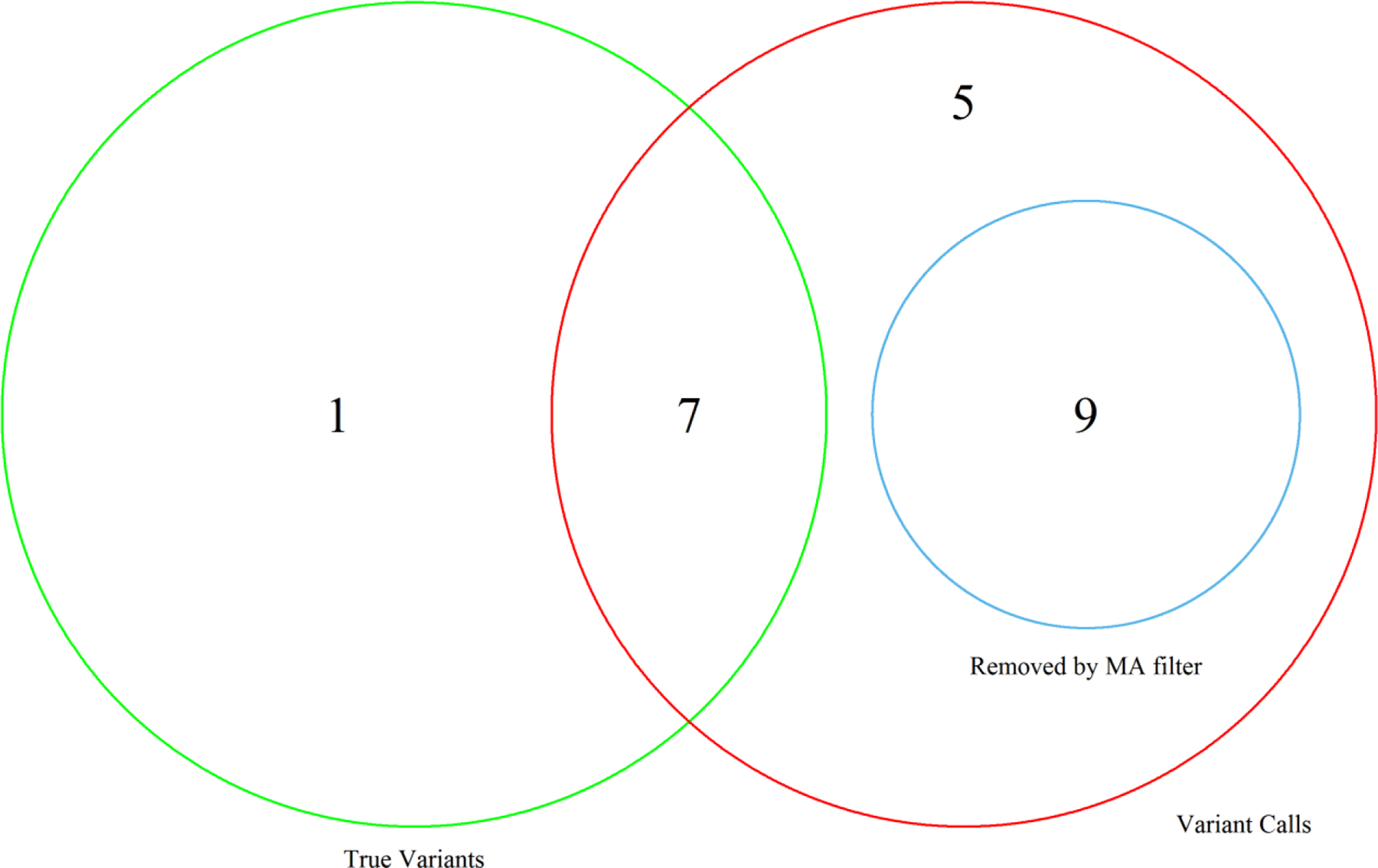
With over 5.5 million deaths worldwide attributed to the respiratory disease COVID-19 caused by the novel coronavirus SARS-CoV-2, it is essential that continued efforts be made to track the evolution and spread of the virus globally. The authors previously presented a rapid and cost-effective method to sequence the entire SARS-CoV-2 genome with 95% coverage and 99.9% accuracy. This method is advantageous for identifying and tracking variants in the SARS-CoV-2 genome compared with traditional short-read sequencing methods which can be time-consuming and costly. Herein, the addition of genotyping probes to a DNA chip that targets known SARS-CoV-2 variants is presented. The incorporation of genotyping probe sets along with the advent of a moving average filter improved the sequencing coverage and accuracy of the SARS-CoV-2 genome.
Keywords: COVID-19; SARS-CoV-2; bioinformatics; genotyping; pandemic; screening; sequencing; tiling-array; viral genome.
Plain language summary
Throughout the COVID-19 pandemic the virus known as SARS-CoV-2 has continued to mutate and evolve. It is imperative to continue to track these mutations and where the virus has traveled to best inform healthcare practices and global strategies to combat the virus. The authors previously developed a method to investigate 95% of this viral genome with 99.9% accuracy that was more cost-effective and less time-consuming than previous methods. In this work, specific markers were added to the technology to allow tracking of mutations in the virus that have already been documented. In doing so, the accuracy and how much of the viral genome can be sequenced was improved.
Conflict of interest statement
The authors declare the following competing financial interest(s): The Centrillion affiliated authors and J.S.E are employees of the company and the company is commercializing the work described herein.
Figures
Update of
-
SARS-CoV-2 Variant Identification Using a Genome Tiling Array and Genotyping Probes.bioRxiv [Preprint]. 2021 May 11:2021.05.11.443659. doi: 10.1101/2021.05.11.443659. bioRxiv. 2021. Update in: Per Med. 2023 Jan;20(1):13-25. doi: 10.2217/pme-2022-0013. PMID: 34013279 Free PMC article. Updated. Preprint.
References
-
- Johns Hopkins University & Medicine. Coronavirus Resource Center (2022). https://coronavirus.jhu.edu/map.html
-
- Drew T The emergence and evolution of swine viral diseases: to what extent have husbandry systems and global trade contributed to their distribution and diversity? Rev Sci Tech 30(1):95–106 (2011). - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous